```{r echo = FALSE}
pacman::p_load(tidyverse, readxl, knitr, kableExtra, Hmisc)
```
# Multinomiale / Ordinale Regression {#sec-multinom-ordinal}
*Letzte Änderung am `r format(fs::file_info("stat-modeling-multinom.qmd")$modification_time, '%d. %B %Y um %H:%M:%S')`*
> *"The amateur does not know what to do. The master knows what not to do." --- James Clear*
Was machen wir wenn wir ein Outcome haben mit mehr als zwei Kategorien. Wenn wir nur zwei Kategorien hätten, dann würden wir eine logistische Regression rechnen. Wenn wir mehr als zwei Kategorien haben, dann sind wir in dem Fall der multinomialen / ordinalen logistischen Regression. Wir rechnen eine multinomialen Regression, wenn wir keine Ordnung in den Kategorien in dem Outcome haben. Wenn wir eine Ordnung vorliegen haben, dann nutzen wir die ordinale Regression. Wir werden uns erstmal eine ordinale Regression anschauen mit nur drei geordneten Stufen. Dann schauen wir uns einmal wie wir eine ordinale Regression auf Boniturnoten in der Likert-Skala rechnen. Wir machen das getrennt, denn wir sind bei wenigen geordneten Kategorien meistens noch am Effekt zwischen den Kategorien interessiert. Im Gegensatz wollen wir bei einem Outcome mit Boniturnoten einen Gruppenvergleich rechnen. Dann interessiert uns der Unterschied und die Effekte zwischen den Boniturnoten nicht. Deshalb trennen wir das hier etwas auf.
Im zweiten Teil wollen wir uns dann noch eine multinominale Regression auf ungeordneten Kategorien eines Outcomes anschauen. Korrekterweise tun wir nur so, als wäre unser vorher geordnetes Outcome dann eben ungeordnet. Das macht dann aber bei deiner Anwendung dann keinen großen Unterschied. Als eine Alternative zur multinationalen Regression stelle ich dann noch die logistische Regression vor. Wir können nämlich einfach unsere Daten nach dem Outcome jeweils in kleinere Datensätze mit nur jeweils zwei der Kategorien aufspalten. Das ist zwar nicht schön, aber auch eine Möglichkeit mit einem Problem umzugehen.
Ich gehe hier nicht auf die Theorie hinter der multinomialen / ordinalen logistischen Regression ein. Wenn dich dazu mehr interessiert findest du in den jeweiligen Abschnitten dann noch eine passende Referenz. Da kannst du dann schauen, welche Informationen du noch zusätzlich findest.
## Annahmen an die Daten
Im folgenden Kapitel zu der multinomialen / ordinalen logistischen linearen Regression gehen wir davon aus, dass die Daten in der vorliegenden Form *ideal* sind. Das heißt wir haben weder fehlende Werte vorliegen, noch haben wir mögliche Ausreißer in den Daten. Auch wollen wir keine Variablen selektieren. Wir nehmen alles was wir haben mit ins Modell. Sollte eine oder mehre Bedingungen nicht zutreffen, dann schaue dir einfach die folgenden Kapitel an.
- Wenn du fehlende Werte in deinen Daten vorliegen hast, dann schaue bitte nochmal in das @sec-missing zu Imputation von fehlenden Werten.
- Wenn du denkst, dass du Ausreißer oder auffälige Werte in deinen Daten hast, dann schaue doch bitte nochmal in das @sec-outlier zu Ausreißer in den Daten.
- Wenn du denkst, dass du zu viele Variablen in deinem Modell hast, dann hilft dir das @sec-variable-selection bei der Variablenselektion.
Daher sieht unser Modell wie folgt aus. Wir haben ein $y$ und $p$-mal $x$. Wobei $p$ für die Anzahl an Variablen auf der rechten Seite des Modells steht. Im Weiteren folgt unser $y$ einer Multinomialverteilung. Damit finden wir im Outcome im Falle der multinomialen logistischen linearen Regression *ungeordnete* Kategorien und im Falle der ordinalen logistischen linearen Regression *geordnete* Kategorien.
$$
y \sim x_1 + x_2 + ... + x_p
$$
Wir können in dem Modell auch Faktoren $f$ haben, aber es geht hier nicht um einen Gruppenvergleich. Das ist ganz wichtig. Wenn du einen Gruppenvergleich rechnen willst, dann musst du in @sec-posthoc nochmal nachlesen.
## Genutzte R Pakete
Wir wollen folgende R Pakete in diesem Kapitel nutzen.
```{r echo = TRUE}
#| message: false
pacman::p_load(tidyverse, magrittr, broom,
parameters, performance, gtsummary,
ordinal, janitor, MASS, nnet, flextable,
emmeans, multcomp, ordinal, see, scales,
janitor, conflicted)
conflicts_prefer(dplyr::select)
conflicts_prefer(dplyr::filter)
conflicts_prefer(magrittr::extract)
```
An der Seite des Kapitels findest du den Link *Quellcode anzeigen*, über den du Zugang zum gesamten R-Code dieses Kapitels erhältst.
## Daten
Im Folgenden wollen wir uns die Daten von den infizierten Ferkeln noch einmal anschauen. Wir nehmen als Outcome die Spalte `frailty` und damit die Gebrechlichkeit der Ferkel. Die Spalte ordnen wir einmal nach *robust*, *pre-frail* und *frail*. Wobei *robust* ein gesundes Ferkel beschreibt und *frail* ein gebrechliches Ferkel. Damit wir später die Richtung des Effekts richtig interpretieren können, müssen wir von *gut* nach *schlecht* sortieren. Das brauchen wir nicht, wenn wir Boniturnoten haben, dazu mehr in einem eigenen Abschnitt. Wir bauen uns dann noch einen Faktor mit ebenfalls der Spalte `frailty` in der wir so tun, als gebe es diese Ordnung nicht. Wir werden dann die ordinale Regression mit dem Outcome `frailty_ord` rechnen und die multinominale Regression dann mit dem Outcome `frailty_fac` durchführen.
```{r}
pig_tbl <- read_excel("data/infected_pigs.xlsx") |>
mutate(frailty_ord = ordered(frailty, levels = c("robust", "pre-frail", "frail")),
frailty_fac = as_factor(frailty)) |>
select(-infected)
```
Schauen wir uns nochmal einen Ausschnitt der Daten in der @tbl-multinom-pigs an.
```{r}
#| echo: false
#| message: false
#| warning: false
#| label: tbl-multinom-pigs
#| tbl-cap: Auszug aus dem Daten zu den kranken Ferkeln.
#| column: page
raw_pig_tbl <- pig_tbl |>
mutate(frailty_ord = as.character(frailty_ord),
frailty_fac = as.character(frailty_fac))
rbind(head(raw_pig_tbl),
rep("...", times = ncol(raw_pig_tbl)),
tail(raw_pig_tbl)) |>
kable(align = "c", "pipe")
```
Das wären dann die Daten, die wir für unsere Modelle dann brauchen. Schauen wir mal was wir jetzt bei der ordinalen Regression herausbekommen.
## Ordinale logistische Regression {#sec-ordinal}
Es gibt sicherlich einiges an Paketen in R um eine ordinale Regression durchzuführen. Ich nutze gerne die Funktion `polr()` aus dem R Paket `{MASS}`. Daneben gibt es auch noch das R Paket `{ordinal}` mit der Funktion `clm()`, die wir dann noch im Anschluss besprechen werden. Ich nutze jetzt erstmal die Funktion `polr()`, da wir hier noch eine externe Referenz haben, die uns noch detailliertere Informationen liefern kann.
::: column-margin
Ich verweise gerne hier auf das tolle Tutorium [Ordinal Logistic Regression \| R Data Analysis Examples](https://stats.oarc.ucla.edu/r/dae/ordinal-logistic-regression/). Hier erfährst du noch mehr über die Analyse der ordinalen logistischen Regression.
:::
Wir schon erwähnt sparen wir usn die mathematischen Details und utzen gleich die Funktion `polr` auf unserem Outcome `frailty`. Wir müssen keine Verteilungsfamilie extra angeben, dass haben wir schon mit der Auswahl der Funktion getan. Die Funktion `polr` kann nur eine ordinale Regression rechnen und wird einen Fehler ausgeben, wenn das Outcome $y$ nicht passt.
```{r}
#| message: false
#| warning: false
ologit_fit <- polr(frailty_ord ~ age + sex + location + activity + crp +
bloodpressure + weight + creatinin,
data = pig_tbl)
```
Schauen wir uns einmal die Ausgabe des Modellfits der ordinalen Regression mit der Funktion `summary()` an. Wir sehen eine Menge Zahlen und das wichtigste für uns ist ja, dass wir zum einen Wissen, dass wir auch die ordinale Regression auf der $link$-Funktion rechnen. Wir erhalten also wieder eine Transformation des Zusammenhangs zurück, wie wir es schon bei der Poisson Regression sowie bei der logistischen Regression hatten.
[Hier gibt es nur die Kurzfassung der *link*-Funktion. @dormann2013parametrische liefert hierzu in Kapitel 7.1.3 nochmal ein Einführung in das Thema.]{.aside}
```{r}
#| message: false
#| warning: false
ologit_fit |> summary()
```
Unsere Ausgabe teilt sich in zwei Teile auf. In dem oberen Teil sehen wir die Koeffizienten des Modells zusammen mit dem Fehler und der Teststatistik. Was wir nicht sehen, ist ein $p$-Wert. Die Funktion rechnet uns keinen Signifikanztest aus. Das können wir aber gleich selber machen. In dem Abschnitt `Intercepts` finden wir die Werte für die Gruppeneinteilung auf der *link*-Funktion wieder. Wir transformieren ja unsere drei Outcomekategorien in einen kontinuierliche Zahlenzusammenhang. Trotzdem müssen ja die drei Gruppen auch wieder auftauchen. In dem Abschnitt `Intercepts` finden wir die Grenzen für die drei Gruppen auf der *link*-Funktion.
::: column-margin
Wir gibt auch ein Tutorial für [How do I interpret the coefficients in an ordinal logistic regression in R?](https://stats.oarc.ucla.edu/r/faq/ologit-coefficients/)
:::
Berechnen wir jetzt einmal die $p$-Werte per Hand. Dafür brauchen wir die absoluten Werte aus der `t value` Spalte aus der `summary` des Modellobjekts. Leider ist die Spalte nicht schön formatiert und so müssen wir uns etwas strecken um die Koeffizienten sauber aufzuarbeiten. Wir erhalten dann das Objekt `coef_tbl` wieder.
```{r}
#| message: false
#| warning: false
coef_tbl <- summary(ologit_fit) |>
coef() |>
as_tibble(rownames = "term") |>
clean_names() |>
mutate(t_value = abs(t_value))
coef_tbl
```
Um die Fläche rechts von dem $t$-Wert zu berechnen, können wir zwei Funktionen nutzen. Die Funktion `pnorm()` nimmt eine Standradnormalverteilung an und die Funktion `pt()` vergleicht zu einer $t$-Verteilung. Wenn wir *rechts* von der Verteilung schauen wollen, dann müssen wir die Option `lower.tail = FALSE` wählen. Da wir auch zweiseitig statistisch Testen, müssen wir den ausgerechneten $p$-Wert mal zwei nehmen. Hier einmal als Beispiel für den $t$-Wert von $1.96$. Mit `pnorm(1.96, lower.tail = FALSE) * 2` erhalten wir $0.05$ als Ausgabe. Das ist unser $p$-Wert. Was uns ja nicht weiter überrascht. Denn rechts neben dem Wert von $1.96$ in einer Standardnormalverteilung ist ja $0.05$. Wenn wir einen $t$-Test rechnen würden, dann müssten wir noch die Freiheitsgrade `df` mit angeben. Mit steigendem $n$ nähert sich die $t$-Verteilung der Standardnormalverteilung an. Wir haben mehr als $n = 400$ Beobachtungen, daher können wir auch `df = 400` setzen. Da kommt es auf eine Zahl nicht an. Wir erhalten mit `pt(1.96, lower.tail = FALSE, df = 400) * 2` dann eine Ausgabe von $0.0507$. Also *fast* den gleichen $p$-Wert.
Im Folgenden setzte ich die Freiheitsgrade `df = 3` dammit wir was sehen. Bei so hohen Fallzahlen wir in unserem beispiel würden wir sonst keine Unterschiede sehen.
```{r}
#| message: false
#| warning: false
coef_tbl |>
mutate(p_n = pnorm(t_value, lower.tail = FALSE) * 2,
p_t = pt(t_value, lower.tail = FALSE, df = 3) * 2) |>
mutate(across(where(is.numeric), round, 3))
```
Damit haben wir einmal händisch uns die $p$-Werte ausgerechnet. Jetzt könnte man sagen, dass ist ja etwas mühselig. Gibt es da nicht auch einen einfacheren Weg? Ja wir können zum einen die Funktion `tidy()` nutzen um die 95% Konfidenzintervalle und die exponierten Effektschätzer aus der ordinalen Regresssion zu erhalten. Wir erhalten aber wieder keine $p$-Werte sondern müssten uns diese $p$- Werte dann wieder selber berechnen.
```{r}
#| message: false
#| warning: false
ologit_fit |>
tidy(conf.int = TRUE, exponentiate = TRUE) |>
select(-coef.type)
```
Um all dieses Berechnen zu umgehen, können wir dann auch die Funktion `model_parameters()` nutzen. Hier berechnen wir dann die $p$-Wert mit $df = 400$ aus einer $t$-Verteilung. Damit umgehen wir das Problem, dass unser Modellfit keine $p$-Werte liefert.
```{r}
#| message: false
#| warning: false
ologit_fit |>
model_parameters()
```
In @tbl-regression-ordinal sehen wir nochmal die Ergebnisse der ordinalen Regression einmal anders aufgearbeitet. Wir aber schon bei der Funktion `tidy()` fehlen in der Tabelle die $p$-Werte. Wir können aber natürlich auch eine Entscheidung über die 95% Konfidenzintervalle treffen. Wenn die 1 mit im 95% Konfidenzintervall ist, dann können wir die Nullhypothese nicht ablehnen.
```{r}
#| message: false
#| warning: false
#| label: tbl-regression-ordinal
#| tbl-cap: "Tabelle der Ergebnisse der ordinalen Regression."
ologit_fit |>
tbl_regression(exponentiate = TRUE) |>
as_flex_table()
```
Wi es im gazen Kapitel schon durchscheint, die Interpreation der $OR$ aus einer ordinalen Regression ist nicht einfach, geschweige den intuitiv. Was wir haben ist der Trend. Wir haben unser Outcome von *robust* zu *frail* sortiert und damit von *gut* nach *schlecht*. Wir können so die Richtung der Variablen in unserem Modell interpretieren. Das heißt, dass männliche Ferkel eher von einer Gebrechlichkeit betroffen sind als weibliche Ferkel. Oder wir sagen, dass ein ansteigender CRP Wert führt zu weniger Gebrechlichkeit. Auf diesem Niveau lassen sich die $OR$ einer ordinalen Regression gut interpretieren.
## Gruppenvergleich {#sec-mult-comp-ord-reg}
In diesem Abschnitt wollen wir Gruppenvergleich mit dem *Cumulative Link Models (CLM)* für ordinale Daten rechnen. Oder andersherum, wir haben Boniturdaten vorliegen und wollen hierfür einen multipen Vergleich rechnen. Mehr zu dem Modell findest du im Tutorium zu [Introduction to Cumulative Link Models (CLM) for Ordinal Data](https://rcompanion.org/handbook/G_01.html). Wir konzentrieren uns hier direkt auf die Auswertung an einem Spieldatensatz. Daran können wir dann einfacher erkennen, was bei einem Vergleich rauskommt, wenn wir wissen was wir reingesteckt haben. Wenn du mehr zu dem Thema lesen willst, dann hilft eventuell auch die Hilfeseite zu [Ordinal models with {emmeans}](https://cran.r-project.org/web/packages/emmeans/vignettes/sophisticated.html#ordinal). Ich muss aber sagen, dass die Seite etwas theoretisch ist.
Wir bauen uns jetzt einen Spieldatensatz mit zwanzig Boniturnoten für drei Tierarten. Ich nehme hier nur fünf Notenschritte, da sonst die Sachlage sehr unübersichtlich wird. Im Prinzip generieren wir uns mit der Funktion `sample` nach vorgegeben Wahrscheinlichkeiten für jede Note zwanzig Boniturnoten für jede Tierart. Dabei haben Katzen eine niedrigere Note als Hunde und die Hunde haben schlechtere Noten als die Füchse. Am Ende brauchen wir dann noch einen geordneten Faktor `likert_ord` damit wir die ordinale Regression rechnen können. Die ursprünglichen Noten behalte ich als numerisch um die Daten besser in `{ggplot}` abbilden zu können.
```{r}
#| message: false
#| warning: false
set.seed(20231201)
n_grp <- 20
grade_tbl <- tibble(trt = gl(3, n_grp, labels = c("cat", "dog", "fox")),
likert = c(sample(1:5, size = n_grp, replace = TRUE, prob = c(0.2, 0.5, 0.2, 0.1, 0.0)),
sample(1:5, size = n_grp, replace = TRUE, prob = c(0.1, 0.2, 0.5, 0.2, 0.0)),
sample(1:5, size = n_grp, replace = TRUE, prob = c(0.0, 0.0, 0.2, 0.5, 0.3)))) |>
mutate(likert_ord = ordered(likert))
```
Dann berechnen wir einmal die mittleren Boniturnoten für die drei Tierarten. Wir sehen, dass wir ziemlich gerade Durchschnittsnoten haben.
```{r}
grade_tbl |>
group_by(trt) |>
summarise(mean(likert))
```
Jetzt können wir die Daten auch schon in die ordinale Regression mit `clm()` stecken. Da die Noten alle den gleichen Abstand zueinander haben, nutzen wir die Option `threshold = "symmetric"`. Wir brauchen hier die Ausgabe der Moelldierung gar nicht weiter im Detail. Wir nutzen den Fit dann gleich in der ANOVA und `{emmeans}` für den Gruppenvergleich der mittleren Boniturnoten.
```{r}
clm_fit <- clm(likert_ord ~ trt, data = grade_tbl,
threshold = "symmetric")
```
Die ANOVA funktioniert wie gewohnt. Wir müssen hier auch nichts anpassen, wir kriegen einfach so unsere p-Werte geliefert. Wir haben einen signifikanten Effekt der Tierarten, was ja auch zu erwarten war. Die Boniturnoten unterscheiden sich zwischen Katzen, Hunden und Füchsen.
```{r}
anova(clm_fit)
```
Dann können wir auch schon den multipen Gruppenvergleich rechnen. Da wir die mittleren Boniturnoten wiedergegeben haben wollen, nutzen wir die Option `mode = "mean.class"`. Sonst würden wir andere Werte erhalten, die dann wirklich schwer zu interpretieren sind. Wie du siehst, sind es die gleichen Werte die wir auch oben für unsere Mittelwerte über die Boniturnoten berechnet haben.
```{r}
emm_obj <- clm_fit |>
emmeans(~ trt, mode = "mean.class")
```
Dann können wir uns auch schon das *compact letter display* wiedergeben lassen. Auch hier gibt es keine Überraschung, die drei Tierarten unterscheiden sich. Mit der Ausgabe könnten wir dann auch gleich ein Säulendigramm erstellen, aber das zeige ich gleich mal in dem zweiten Beispiel für zwei Faktoren und der Bonitur von Weizen.
```{r}
emm_obj |>
cld(Letters = letters)
```
In unserem zweiten Beispiel schauen wir uns einen zweifaktoriellen Datensatz einmal an. Wir haben Weizen angepflanzt und bonitieren die Weizenpflanzen nach der Likert Skala. Dabei bedeutet dann eine 1 ein schlechte Note und eine 9 die bestmögliche Note. Wir hätten natürlich hier auch einen Kurskal-Wallis-Test rechnen können und dann im Anschluss einen paarweisen Wilcoxon Test. Nun modellieren wir hier aber die Boniturnoten mal mit einer ordinalen Regression und rechnen den anschließenden Gruppenvergleich dann mit dem R Paket `{emmeans}`.
Unser Datensatz `grade_tbl` enthält den Faktor `block` mit drei Levels sowie den Faktor `variety` mit fünf Leveln. Jedes Level repräsentiert dabei eine Weizensorte. Wichtig ist hier, dass wir die Noten als *geordneten* Faktor mit der Funktion `ordered` erstellen. Nur dann haben die Noten eine Ordnung und die folgenden Funktionen erkennen dann auch die Spalte `grade_ord` als eine ordinale Spalte mit Noten.
```{r}
#| message: false
#| warning: false
grade_tbl <- tibble(block = rep(c("I", "II", "III"), each = 3),
A = c(2, 3, 4, 3, 3, 2, 4, 2, 1),
B = c(7, 9, 8, 9, 7, 8, 9, 6, 7),
C = c(6, 5, 5, 7, 5, 6, 4, 7, 6),
D = c(2, 3, 1, 2, 1, 1, 2, 2, 1),
E = c(4, 3, 7, 5, 6, 4, 5, 7, 5)) |>
gather(key = variety, value = grade, A:E) |>
mutate(grade_ord = ordered(grade))
```
Wir schauen uns nochmal den Datensatz an und sehen, dass wir einmal die Spalte `grade` als numerische Spalte vorliegen haben und einmal als geordneten Faktor. Wir brauchen die numerische Spalte um die Daten besser in `ggplot()` darstellen zu können.
```{r}
grade_tbl |> head(4)
```
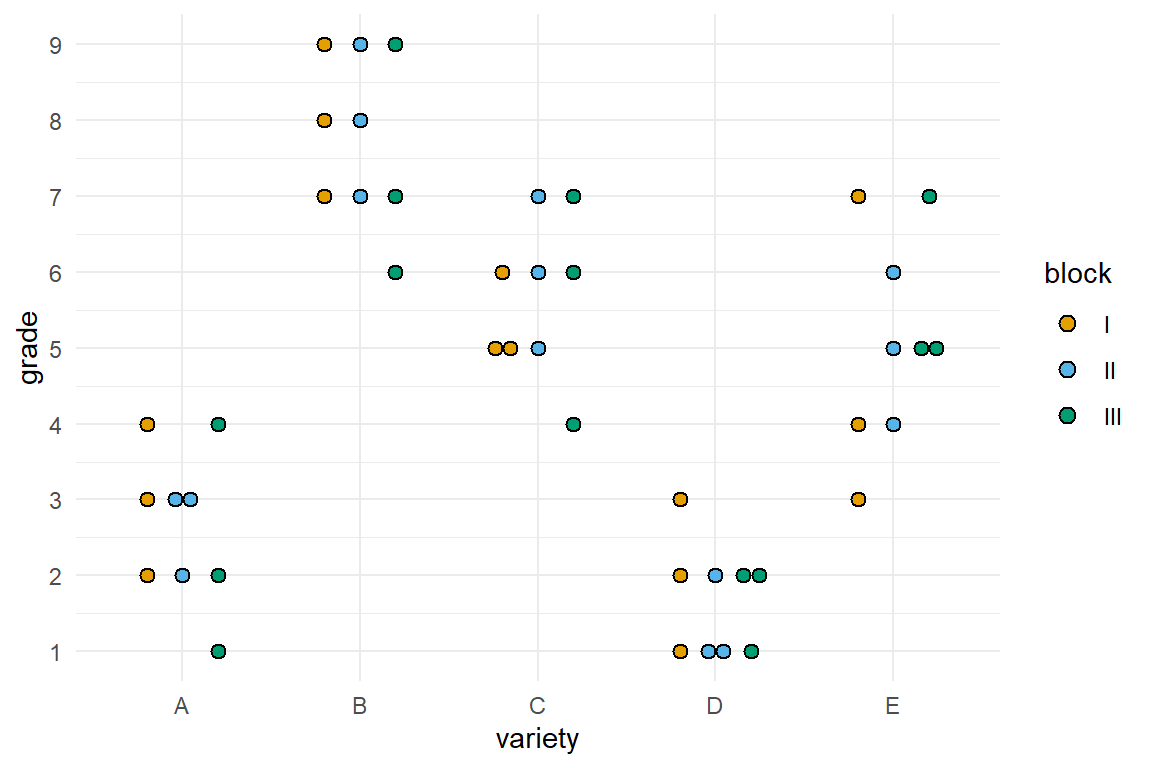
In @fig-clm-dot sehen wir einmal die Daten als Dotplot dargestellt. Auf der x-Achse sind die Weizensorten und auf der y-Achse die Boniturnoten. Ich habe noch die zusätzlichen Linien für jede einzelne Note mit eingezeichnet.
```{r}
#| echo: true
#| message: false
#| label: fig-clm-dot
#| fig-align: center
#| fig-height: 4
#| fig-width: 6
#| fig-cap: "Dotplot des Datenbeispiels für die Bonitur von fünf Weizensorten."
ggplot(grade_tbl, aes(variety, grade, fill = block)) +
theme_minimal() +
geom_dotplot(binaxis = "y", stackdir='center',
position=position_dodge(0.6), dotsize = 0.75) +
scale_y_continuous(breaks = 1:9, limits = c(1,9)) +
scale_fill_okabeito()
```
Jetzt können wir schon die Funktion `clm()` aus dem R Paket `{ordinal}` verwenden um die ordinale Regression zu rechnen. Wir haben in dem R Paket `{ordinal}` noch weitere Modelle zu Verfügung mit denen wir auch komplexere Designs bis hin zu linearen gemischten Modellen für eine ordinale Regresssion rechnen können. Da wir mit Boniturnoten als Outcome arbeiten setzen wir auch die Option `threshold = "symmetric"`. Damit teilen wir der Funktion `clm()` mit, dass wir es mit einer symmetrischen Notenskala zu tun haben. Wenn du das nicht hast, dass kannst du die Option auch auf `"flexible"` stellen. Dann wird eine nicht symmetrische Verteilung des Outcomes angenommen.
```{r}
#| message: false
#| warning: false
clm_fit <- clm(grade_ord ~ variety + block + variety:block, data = grade_tbl,
threshold = "symmetric")
```
Es ist auch möglich auf dem Modellfit eine ANOVA zu rechnen. Wir machen das hier einmal, aber wir erwarten natürlich einen signifikanten Effekt von der Sorte. Die Signifikanz konnten wir ja schon oben im Dotplot sehen.
```{r}
#| message: false
#| warning: false
anova(clm_fit)
```
Jetzt nutzen wir wieder den Modellfit für unseren Gruppenvergleich in `{emmeans}`. Wir nutzen dafür wieder die Funktion `emmeans()` und lassen uns das *compact letter display* über die Funktion `cld()` wiedergeben. Da wir hier eigentlich keinen signifikanten Effekt der Blöcke vorliegen haben, könnten wir auch einfach die Option `~ variety` nutzen und den Block weglassen. Dann wäre auch die Ausgabe mit den *compact letter display* etwas übersichtlicher. Ich habe dann noch die Ausgabe einmal nach den Sorten sortiert und nicht nach dem *compact letter display* um hier etwas mehr Übersicht zu erhalten.
```{r}
#| message: false
#| warning: false
emm_obj <- clm_fit |>
emmeans(~ variety * block, mode = "mean.class") |>
cld(Letters = letters) |>
arrange(variety)
emm_obj
```
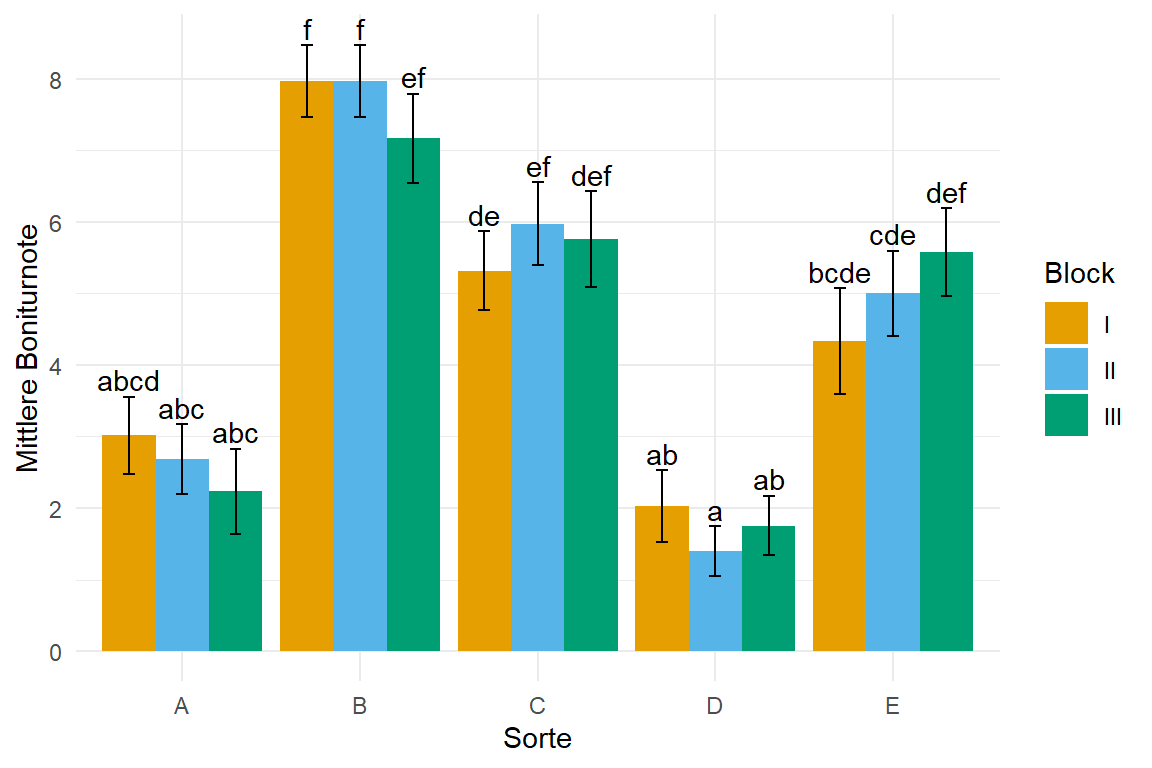
In der @fig-log-mod-ordinal-weizen siehst du dann einmal die Ausgabe des multipen Gruppenvergleichs visualisiert. Hier wäre es dann schon fast sinnvoll über die Blöcke zu mitteln und daher den Block aus dem Modell zu nehmen. Der Block hat keinen Effekt und sorgt aber für noch mehr Vergleiche, die gerechnet werden müssen. Du kannst dann ja oben nochmal die Funktion `emmeans()` anpassen und gemittelt über alle Blöcke mit der Option `~ variety` rechnen. Die Funktion `str_trim()` entfernt dann noch die Leerzeichen von dem *compact letter display* und zentriert damit alles schön.
```{r}
#| echo: true
#| warning: false
#| message: false
#| label: fig-log-mod-ordinal-weizen
#| fig-align: center
#| fig-height: 4
#| fig-width: 6
#| fig-cap: "Säulendigramm der mittleren Boniturnote des Weizen aus einer ordinalen Regression. Das *compact letter display* wird dann in `{emmeans}` generiert. Teilweise kommt das *compact letter display* an seine visuellen Grenzen. Hier wäre es dann schon fast sinnvoll über die Blöcke zu mitteln und daher den Block aus dem Modell zu nehmen."
emm_obj |>
as_tibble() |>
ggplot(aes(x = variety, y = mean.class, fill = block)) +
theme_minimal() +
labs(y = "Mittlere Boniturnote", x = "Sorte",
fill = "Block") +
geom_bar(stat = "identity",
position = position_dodge(width = 0.9, preserve = "single")) +
geom_text(aes(label = str_trim(.group), y = mean.class + SE + 0.01),
position = position_dodge(width = 0.9), vjust = -0.25) +
geom_errorbar(aes(ymin = mean.class-SE, ymax = mean.class+SE),
width = 0.2,
position = position_dodge(width = 0.9, preserve = "single")) +
scale_fill_okabeito()
```
::: callout-tip
## Anwendungsbeispiel: Gruppenvergleich für eine Bonitur
Im folgenden Beispiel schauen wir uns nochmal ein praktische Auswertung von einem agrarwissenschaftlichen Beispiel mit Brokkoli an. Wir haben uns in diesem Experiment verschiedene Dosen `fert_amount` von einem Dünger aufgebracht sowie verschiedene Zeitpunkte der Düngung `fert_time` berücksichtigt. Ziel ist es die Boniturnoten für den Stamm von Brokkoli miteinander zu vergleichen. Auch hier haben wir einige Besonderheiten in den Daten, da nicht jede Faktorkombination vorliegt. Wir ignorieren aber diese Probleme und rechnen einfach stumpf unseren Gruppenvergleich. Wir müssen aber ein paar Anpassungen durchführen. Unsere Noten müssen ein geordneter Faktor sein, daher nutzen wir die Funktion `ordered`. Darüber hinaus schmeißen wir die Düngerzeit `early` aus den Daten, da wir zu dem Zeitpunkt keine hohlen Stämme bonitiert haben.
```{r}
broc_tbl <- read_excel("data/broccoli_weight.xlsx") |>
mutate(fert_time = factor(fert_time, levels = c("none", "early", "late")),
fert_amount = as_factor(fert_amount),
block = as_factor(block),
stem_hollowness_num = stem_hollowness,
stem_hollowness = ordered(stem_hollowness)) |>
filter(fert_time == "early") |>
select(fert_time, fert_amount, block, stem_hollowness, stem_hollowness_num) |>
droplevels() |>
na.omit()
```
Im Folgenden einmal die Tabelle mit `tabyl` aus dem R Paket `{janitor}`. Mehr dazu auf der Hilfeseite zu [tabyls: a tidy, fully-featured approach to counting things](https://cran.r-project.org/web/packages/janitor/vignettes/tabyls.html). Wir sehen, dass wir kaum schlechte Noten erhalten. Fast alle Brokkoliköpfe haben eine 1 erhalten, was für intakte Köpfe ohne einen hohlen Stamm spricht.
```{r}
broc_tbl |>
tabyl(stem_hollowness, fert_amount)
```
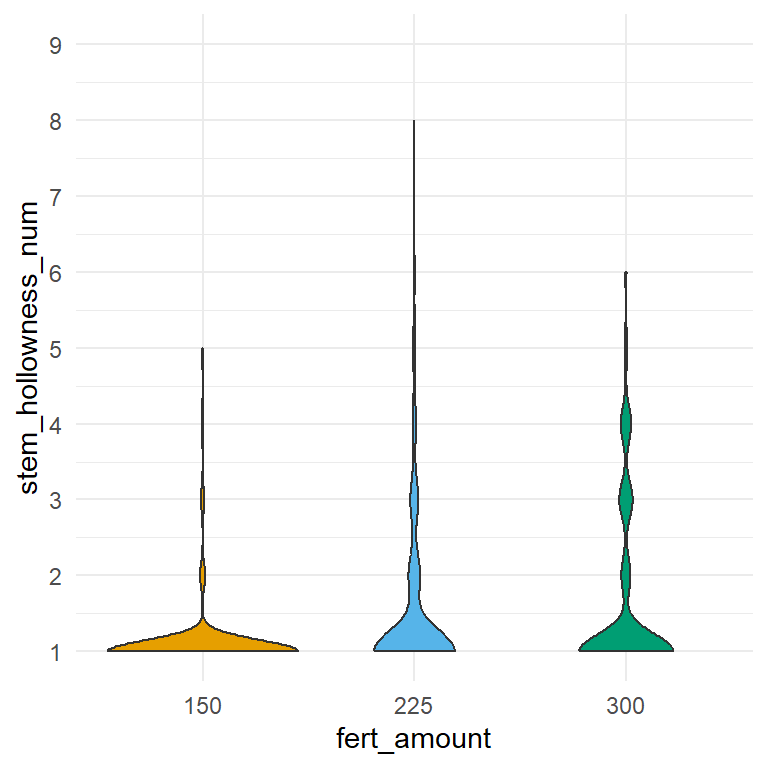
Die Tabelle sehen wir dann auch nochmal in der @fig-clm-broc-violin als Violinplot visualisiert. Wir sehen hier nochmal sehr drastisch, dass wir kaum Noten großer als Eins in den Daten vorliegen haben.
```{r}
#| echo: true
#| message: false
#| label: fig-clm-broc-violin
#| fig-align: center
#| fig-height: 4
#| fig-width: 4
#| fig-cap: "Violinplot der Boniturnoten der Brokkolistämme. Klar ist die sehr schiefe Verteilung der Boniturnoten zu erkennen."
ggplot(broc_tbl, aes(fert_amount, stem_hollowness_num, fill = fert_amount)) +
theme_minimal() +
geom_violin() +
scale_y_continuous(breaks = 1:9, limits = c(1,9)) +
scale_fill_okabeito() +
theme(legend.position = "none")
```
Wir rechnen jetzt natürlich trotzdem eine ordinale Regression mit der Funktion `clm()`. Wir setzen die Option `theshold = "symmetric"` da wir davon ausgehen, dass unsere Noten alle den gleichen Abstand zueinander haben.
```{r}
clm_fit <- clm(stem_hollowness ~ fert_amount,
data = broc_tbl, threshold = "symmetric")
```
Jetzt rechnen wir in den beiden folgenden Tabs einmal die ANOVA und dann auch den multiplen Gruppenvergleich mit `{emmeans}`. Da wir hier ordinale Daten haben, können wir dann nicht einfach die Standardverfahren nehmen. Wir entscheiden uns dann für den Standardfehler bei der Darstellung.
::: panel-tabset
## ANOVA mit `anova()`
Wir rechnen hier einmal die ANOVA und nutzen den $\mathcal{X}^2$-Test für die Ermittelung der p-Werte. Wir müssen hier einen Test auswählen, da per Standardeinstellung kein Test gerechnet wird. Wir machen dann die Ausgabe nochmal schöner und fertig sind wir.
```{r}
clm_fit |>
anova() |>
model_parameters()
```
Wir sehen, dass der die Menge des Düngers signifikant ist. Wir haben nicht mehr Faktoren in dem Modell gehabt, so dass wir hier auch keine weiteren Aussagen tätigen können.
## Gruppenvergleich mit `emmeans()`
Im Folgenden rechnen wir einmal für den Faktor `fert_amount` einen Gruppenvergleich. Wir setzen hier die Option `mean.class` damit wir dann die mittleren Noten wiedergegeben bekommen. Mit den mittleren Noten können wir dann ein Säulendiagramm erstellen. Dann adjustieren wir noch nach Bonferroni und sind fertig.
```{r}
emm_obj <- clm_fit |>
emmeans(~ fert_amount, mode = "mean.class") |>
cld(Letters = letters, adjust = "bonferroni")
emm_obj
```
Das `emm_obj` Objekt werden wir dann gleich einmal in `{ggplot}` visualisieren. Die `mean.class` stellt den mittleren Noten des Brokkoli für die Menge der Düngung dar. Wir nutzen dann auch hier die Standardfehler für die Abbildungen, da wir sonst schnelle Werte kleiner 0 und größer 9 erhalten. Dann können wir zum Abschluss auch das *compact letter display* anhand der Abbildung interpretieren.
Gerade in diesem Beispiel bietet es sich an, dass wir explizit *mindestens* einen Noten*punkt*unterschied vorliegen haben wollen, damit wir von einem relevanten Unterschied sprechen können. Die Funktion `cld()` erlaubt es ein $\Delta$ zu definieren, dass mindestens überschritten sein muss, damit wir einen Unterschied feststellen. Alles was in dem Bereich $\pm\Delta$ liegt, gilt dann als gleich auch wenn es sonst signifikant wäre. Deshalb setzen wir die Option `delta = 1` um sicherzustellen, dass nur *relevante* Unterschiede auch als solche angezeigt werden. Daher haben wir nach der Anpassung auch keine signifikanten Unterschiede mehr. Alle Gruppen sind gleich.
```{r}
clm_fit |>
emmeans(~ fert_amount, mode = "mean.class") |>
cld(Letters = letters, adjust = "bonferroni", delta = 1)
```
:::
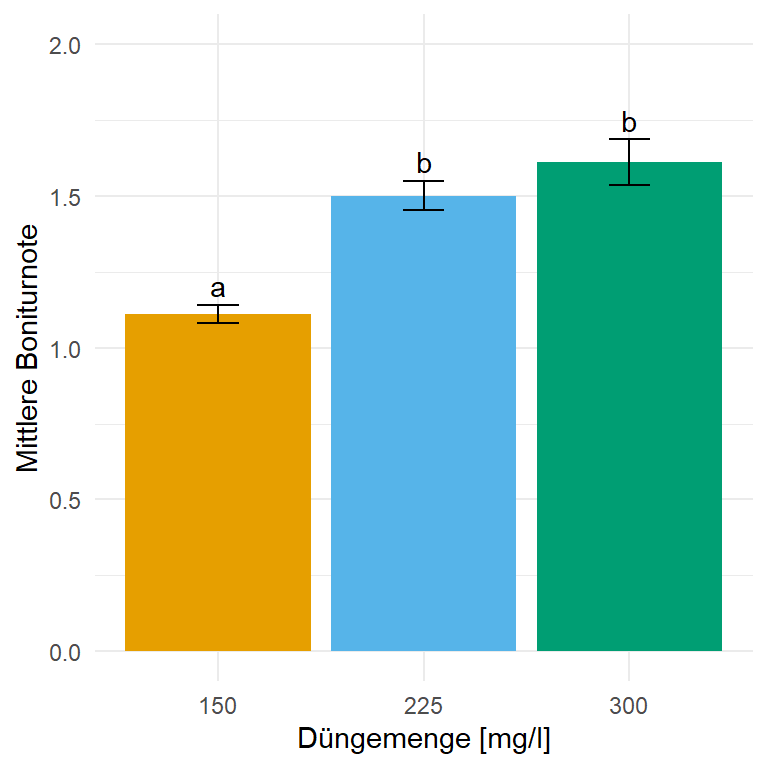
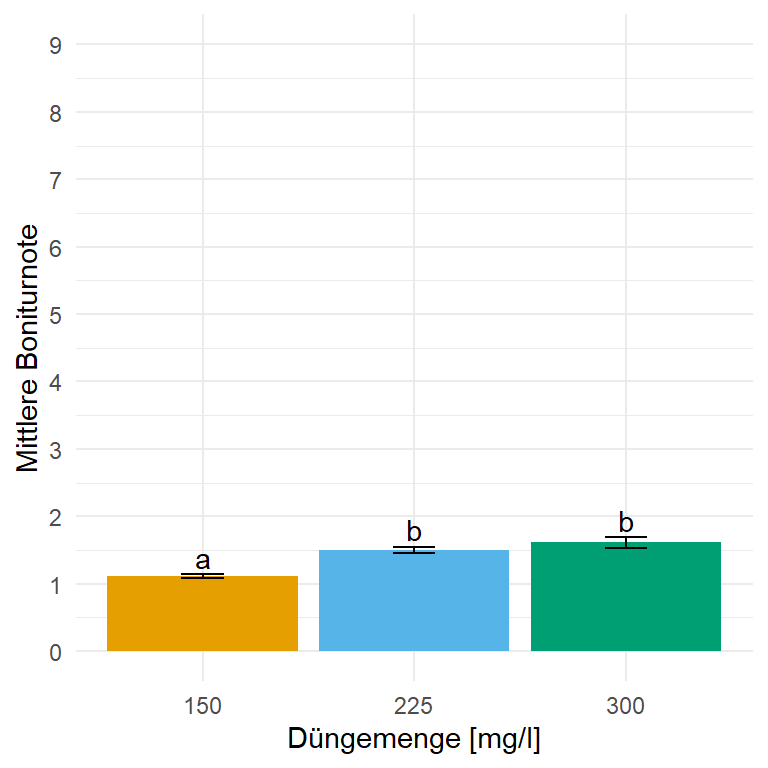
In der @fig-log-mod-ordinal-broc siehst du das Ergebnis der Auswertung in einem Säulendiagramm. Zwar sind die mittleren Boniturnoten signifikant unterschiedlich, aber ist der Effekt auf der vollen Boniturnotenskala kaum zu sehen. Wir haben es hier mit knapp einer Notenstufe Unterschied zu tun und das ist wirklich wenig. Daher wäre hier mal der Fall, dass wir einen signifikanten aber nicht relevanten Unterschied vorliegen haben.
```{r}
#| echo: true
#| warning: false
#| message: false
#| label: fig-log-mod-ordinal-broc
#| fig-align: center
#| fig-height: 4
#| fig-width: 4
#| fig-cap: "Säulendigramm der mittleren Boniturnote des Brokkoli aus einer ordinalen Regression. Das `clm()`-Modell berechnet das mittlere Boniturnote für den Faktor `fert_amount`. Das *compact letter display* wird dann in `{emmeans}` generiert. Wir nutzen hier den Standardfehler, da die Standardabweichung mit der großen Fallzahl rießig wäre."
#| fig-subcap:
#| - "Reduzierte Boniturskala."
#| - "Volle Boniturskala."
#| layout-nrow: 1
emm_obj |>
as_tibble() |>
ggplot(aes(x = fert_amount, y = mean.class, fill = fert_amount)) +
theme_minimal() +
labs(y = "Mittlere Boniturnote", x = "Düngemenge [mg/l]",
fill = "Düngemenge [mg/l]") +
geom_bar(stat = "identity") +
geom_text(aes(label = str_trim(.group), y = mean.class + SE + 0.01), vjust = -0.25) +
geom_errorbar(aes(ymin = mean.class-SE, ymax = mean.class+SE),
width = 0.2) +
scale_fill_okabeito() +
theme(legend.position = "none") +
scale_y_continuous(breaks = c(0, 0.5, 1.0, 1.5, 2.0), limits = c(0, 2))
emm_obj |>
as_tibble() |>
ggplot(aes(x = fert_amount, y = mean.class, fill = fert_amount)) +
theme_minimal() +
labs(y = "Mittlere Boniturnote", x = "Düngemenge [mg/l]",
fill = "Düngemenge [mg/l]") +
geom_bar(stat = "identity") +
geom_text(aes(label = str_trim(.group), y = mean.class + SE + 0.01), vjust = -0.25) +
geom_errorbar(aes(ymin = mean.class-SE, ymax = mean.class+SE),
width = 0.2) +
scale_fill_okabeito() +
theme(legend.position = "none") +
scale_y_continuous(breaks = 0:9, limits = c(0,9))
```
:::
## Multinomiale logistische Regression {#sec-multinom}
Was machen wir in eine multinomialen logistische Regression? Im Gegensatz zu der ordinalen Regression haben wir in der multinominalen Regression keine Ordnung in unserem Outcome. Das macht die Sache dann schon eine Nummer komplizierter. Und wir lösen dieses Problem indem wir ein Level des Outcomes oder eben eine Kategorie des Outcomes als *Referenz* definieren. Dann haben wir wieder unsere Ordnung drin. Und die Definition der Referenz ist auch manchmal das schwerste Unterfangen. Wenn ich keine Ordnung in meinem Outcome habe, wie soll ich dann die Referenz bestimmen? Aber das ist dann immer eine Frage an den konkreten Datensatz. Hier basteln wir uns ja die Fragestellung so hin, dass es passt.
::: column-margin
Ich verweise gerne hier auf das tolle Tutorium [Multinomial Logistic Regression \| R Data Analysis Examples](https://stats.oarc.ucla.edu/r/dae/multinomial-logistic-regression/). Hier erfährst du noch mehr über die Analyse der multinominale logistischen Regression.
:::
Um eine Referenz in dem Outcome zu definieren nutzen wir die Funktion `relevel()` und setzen als unsere Referenz das Level `frail` aus unserem Outcome `frailty`. Wir hätten auch jedes andere Level als Referenz nehmen können. Zu dieser Referenz werden wir jetzt unser Modell anpassen. Ich nehme immer als Referenz das *schlechteste* im Sinne von *nicht gut*. In unserem Fall ist das eben das Level `frail`.
```{r}
#| message: false
#| warning: false
pig_tbl <- pig_tbl |>
mutate(frailty_fac = relevel(frailty_fac, ref = "frail"))
```
Nachdem wir unsere Referenz definiert haben, können wir wieder recht einfach mit der Funktion `multinom()` aus dem Paket `{nnet}` die multinominalen Regression rechnen. Ich mache keinen Hehl daraus. Ich mag die Funktion nicht, da die Ausgabe der Funktion sehr unsortiert ist und uns nicht gerade die Arbeit erleichtert. Auch schweigt die Funktion nicht, sondern muss immer eine Ausgabe wiedergeben. Finde ich sehr unschön.
```{r}
#| message: false
#| warning: false
multinom_fit <- multinom(frailty_fac ~ age + sex + location + activity + crp + bloodpressure + weight + creatinin,
data = pig_tbl)
```
Die Standardausgabe von `multinom()` hat wiederum keine $p$-Werte und wir könnten uns über die Funktion `pnorm()` wiederum aus den $t$-Werten unsere $p$-Werte berechnen. Leider erspart sich `multinom()` selbst den Schritt die $t$-Werte zu berechnen, so dass wir die $t$-Werte selber berechnen müssen. Nicht das es ein Problem wäre, aber schön ist das alles nicht. Im Folgenden siehst du dann einmal die Berechnung der $p$-Werte über die Berechnung der Teststatistik.
```{r}
#| message: false
#| warning: false
z_mat <- summary(multinom_fit)$coefficients/summary(multinom_fit)$standard.errors
p_n <- (1 - pnorm(abs(z_mat), 0, 1)) * 2
p_n
```
Jetzt müssten wir diese $pp$-Werte aus der Matrix noch mit unseren Koeffizienten verbauen und da hört es dann bei mir auf. Insbesondere da wir ja mit `model_parameters()` eine Funktion haben, die uns in diesem Fall wirklich gut helfen kann. Wir nehmen hier zwar die $t$-Verteilung an und haben damit leicht höre $p$-Werte, aber da wir eine so große Anzahl an Beobachtungen haben, fällt dieser Unterschied nicht ins Gewicht.
```{r}
#| message: false
#| warning: false
multinom_fit |> model_parameters(exponentiate = TRUE)
```
Was sehen wir? Zuerst haben wir etwas Glück. Den unsere Referenzlevel macht dann doch Sinn. Wir vergleichen ja das Outcomelevel `robust` zu `frail` und das Outcomelevel `pre-frail` zu `frail`. Dann haben wir noch das Glück, dass durch unsere Ordnung dann auch `frail` das schlechtere Outcome ist, so dass wir die $OR$ als Risiko oder als protektiv interpretieren können. Nehmen wir als Beispiel einmal die Variable `crp`. Der CRP Wert höht das Risiko für `frail`. Das macht schonmal so Sinn. Und zum anderen ist der Effekt bei dem Vergleich von `pre-frail` zu `frail` mit $1.16$ nicht so große wie bei `robust` zu `frail` mit $1.26$. Das macht auch Sinn. Deshalb passt es hier einigermaßen.
In @tbl-tbl-regression-multinom sehen wir nochmal die Ausgabe von einer multinominalen Regression durch die Funktion `tbl_regression()` aufgearbeitet.
```{r}
#| message: false
#| warning: false
#| label: tbl-tbl-regression-multinom
#| tbl-cap: "Tabelle der Ergebnisse der multinominalen Regression."
multinom_fit |>
tbl_regression(exponentiate = TRUE) |>
as_flex_table()
```
Leider wird die Sache mit einer multinominalen Regression sehr unangenehm, wenn wir wirklich nicht sortierbare Level im Outcome haben. Dann haben wir aber noch ein Möglichkeit der multinominalen Regression zu entkommen. Wir rechnen einfach separate logistische Regressionen. Die logistischen Regressionen können wir dann ja separat gut interpretieren.
## Logistische Regression als Ausweg
::: callout-important
## Bitte Beachten bei der Berechung über separate logistische Regressionen
Durch die Verwendung von separaten logistischen Regressionen vermindern wir die Fallzahl je gerechneter Regression, so dass wir *größere* $p$-Werte erhalten werden als in einer multinominalen Regression. Oder andersherum, durch die verminderte Fallzahl in den separaten logistischen Regressionen haben wir eine geringere Power einen signifikanten Unterschied nachzuweisen.
:::
Es gibt den einen Ring um sich zu knechten. Und das ist die logistische Regression. Gut die logistische Regression hilft jetzt nicht, wenn es mit Boniturnoten zu tun hast, aber wenn wir wenige Level im Outcome haben. In unserem Fall haben wir ja drei Level vorliegen, da können wir dann jeweils ein Level rausschmeißen und haben dann nur noch ein binäres Outcome. Das ist auch die zentrale Idee. Wir entfernen immer alle Level bis wir nur noch zwei Level in unserem Outcome haben und rechnen für diese beiden Level dann eine logistische Regression.
Schauen wir uns erstmal an, wie sich die Daten über die drei Kategorien in unserem Outcome verteilen. Wenn wir eine Kategorie im Outcome kaum vorliegen haben, könnten wir diese Daten vielleicht mit einer anderen Kategorie zusammenlegen oder aber müssen von unserer Idee hier Abstand nehmen.
```{r}
#| message: false
#| warning: false
pig_tbl$frailty_fac |> tabyl()
```
Wir haben nicht so viele Beobachtungen in der Kategorie `frail`. Wir könnten also auch die beiden Faktorlevel `pre-frail` und `frail` zusammenlegen. Das R Paket `{forcats}` liefert sehr viele Funktion, die dir helfen Faktoren zu kodieren und zu ändern.
```{r}
pig_tbl$frailty_fac |>
fct_recode(frail_pre_frail = "frail", frail_pre_frail = "pre-frail") |>
tabyl()
```
Das ist jetzt aber nur eine Demonstration für die Zusammenlegung. Wir wollen jetzt trotzdem unsere drei logistischen Regressionen rechnen. Warum drei? Wir haben ja drei Level in unserem Outcome und wir werden jetzt uns drei Datensätze so bauen, dass in jdem Datensatz unser Outcome immer nur zwei Level hat. Die einzelnen Datensätze speichern wir dann in einer Liste.
```{r}
#| message: false
#| warning: false
pig_lst <- list(robust_prefrail = filter(pig_tbl, frailty_fac %in% c("robust", "pre-frail")),
robust_frail = filter(pig_tbl, frailty_fac %in% c("robust", "frail")),
prefrail_frail = filter(pig_tbl, frailty_fac %in% c("pre-frail", "frail")))
```
Wir können das auch fancy. Und das demonstriere ich dann mal hier. Wenn wir die Funktion `combn()` nutzen erhalten wir eine Liste mit allen zweier Kombinationen wieder. Diese Liste können wir dann in die Funktion `map()` stecken, die dann über die Liste unserer Kombinationen iteriert. Pro Liste filtern `map()` dann den Datensatz für uns heraus. Ja, ist ein wenig *over the top*, aber ich wollte das mal für mich mit `map()` ausprobieren und es passte hier so schön.
```{r}
#| message: false
#| warning: false
pig_fancy_lst <- combn(c("robust", "pre-frail", "frail"), 2, simplify = FALSE) |>
map(~filter(pig_tbl, frailty_fac %in% .x))
```
Egal wie du auf die Liste gekommen bist, wir müssen noch die überflüssigen Level *droppen*. Keine Ahnung was das deutsche Wort ist. Vermutlich ist das deutsche Wort dann entfernen. Dann können wir für jeden der Listeneinträge die logistische Regression rechnen. Am Ende lassen wir uns noch die exponierten Modellfits ausgeben. In der letzten Zeile entferne ich noch den Intercept von der Ausgabe des Modells. Den Intercept brauchen wir nun wirklich nicht.
```{r}
#| message: false
#| warning: false
pig_lst |>
map(~mutate(.x, frailty_fac = fct_drop(frailty_fac))) |>
map(~glm(frailty_fac ~ age + sex + location + activity + crp + bloodpressure + weight + creatinin,
data = .x, family = binomial)) |>
map(model_parameters, exponentiate = TRUE) |>
map(extract, -1, )
```
Eine Sache ist super wichtig zu wissen. Wie oben schon geschrieben, durch die Verwendung von separaten logistischen Regressionen vermindern wir die Fallzahl je Regression, so dass wir größere $p$-Werte erhalten werden, als in einer multinominalen Regression. Das ist der Preis, den wir dafür bezahlen müssen, dass wir besser zu interpretierende Koeffizienten erhalten. Und das ist auch vollkommen in Ordnung. Ich selber habe lieber Koeffizienten, die ich interpretieren kann, als unklare Effekte mit niedrigen $p$-Werten.
Schauen wir einmal auf unseren Goldstandard, der Variable für den CRP-Wert. Die Variable haben wir ja jetzt immer mal wieder in diesem Kapitel interpretiert und uns angeschaut. Die Variable `crp` passt von dem Effekt jedenfalls gut in den Kontext mit rein. Die Effekte sind ähnlich wie in der multinominalen Regression. Wir haben eben nur größere $p$-Werte. Jetzt müssen wir entscheiden, wir können vermutlich die getrennten logistischen Regressionen besser beschreiben und interpretieren. Das ist *besonders* der Fall, wenn wir wirklich Probleme haben eine Referenz in der multinominalen Regression festzulegen. Dann würde ich immer zu den getrennten logistischen Regressionen greifen als eine schief interpretierte multinominale Regression.
## Referenzen {.unnumbered}