R Code [zeigen / verbergen]
y_jump_dog <- c(5.7, 8.9, 11.8, 5.6, 9.1, 8.2, 7.6)
y_jump_dog[1] 5.7 8.9 11.8 5.6 9.1 8.2 7.6Letzte Änderung am 06. August 2025 um 20:43:21
“I am fond of pigs. Dogs look up to us. Cats look down on us. Pigs treat us as equals.” — Winston S. Churchill

Die ursprüngliche Idee zu den Sprungweiten der Hunde- und Katzenflöhe stammt von Cadiergues et al. (2000) aus der entsprechenden Veröffentlichung A comparison of jump performances of the dog flea, Ctenocephalides canis (Curtis, 1826) and the cat flea, Ctenocephalides felis felis (Bouché, 1835). Wir bauen uns hier kleinere Datensätze mit einer ähnlichen Fragestellung nach, die wir dann in den folgenden Kapiteln immer wieder nutzen. Dabei begleiten uns die Hunde- und Katzenflöhe eher in den Einführungskapiteln. Später schauen wir uns dann noch komplexere Datensätze mit einem klaren Anwendungsbezug an.
In unserem ersten Beispiel wollen wir uns verschiedene Daten von Hundeflöhen (eng. dog flea) anschauen. Unter anderem sind dies die Sprungweite eines Hundeflohs, die Anzahl an Haaren am rechten Bein eines Hundeflohs, die Boniturnoten auf einer Flohmesse sowie der Infektionsstatus mit Flohschnupfen. Hier nochmal detailliert dargestellt, was wir uns im Folgenden immer wieder anschauen wollen.
Erst einmal die Zahlen, so wie du die Messwerte dann auf einen karierten Zettel aus deinem Collegeblock schreiben würdest. Ich empfehle immer Papier auf dem Feld, Gewächshaus oder Stall. Ein Pad geht da mal schnell kaputt. Dann habe ich zu deinen Notizen noch die mathematischen Schreibweise sowie die Schreibweise in R ergänzt. In R nutze ich auch schon gleich ein Objekt um die Zahlen zu speichern.
Sprungweite in [cm] von verschiedenen Flöhen:

\[ Y_{jump} = \{5.7, 8.9, 11.8, 5.6, 9.1, 8.2, 7.6\}. \]
y_jump_dog <- c(5.7, 8.9, 11.8, 5.6, 9.1, 8.2, 7.6)
y_jump_dog[1] 5.7 8.9 11.8 5.6 9.1 8.2 7.6Anzahl an Haaren am rechten Bein von verschiedenen Flöhen:

\[ Y_{count} = \{18, 22, 17, 12, 23, 18, 21\}. \]
y_count_dog <- c(18, 22, 17, 12, 23, 18, 21)
y_count_dog[1] 18 22 17 12 23 18 21Gewicht des gesprungenen Flohes in [mg] von verschiedenen Flöhen:

\[ Y_{weight} = \{2.1, 2.3, 2.8, 2.4, 1.2, 4.1, 3.2\}. \]
y_weight_dog <- c(2.1, 2.3, 2.8, 2.4, 1.2, 4.1, 3.2)
y_weight_dog[1] 2.1 2.3 2.8 2.4 1.2 4.1 3.2Boniturnoten [1 = schwächste bis 9 = stärkste Ausprägung] von verschiedenen Flöhen:

\[ Y_{grade} = \{8, 8, 6, 8, 7, 7, 9\}. \]
y_grade_dog <- c(8, 8, 6, 8, 7, 7, 9)
y_grade_dog[1] 8 8 6 8 7 7 9Infektionstatus [0 = gesund, 1 = infiziert] mit Flohschnupfen von verschiedenen Flöhen:

\[ Y_{infected} = \{0, 1, 1, 0, 1, 0, 0\}. \]
y_infected_dog <- c(0, 1, 1, 0, 1, 0, 0)
y_infected_dog[1] 0 1 1 0 1 0 0Je nachdem was wir messen, nimmt \(Y\) andere Zahlenräume an. Wir bezeichnen unser gemessenes \(Y\) auch gerne als Messwert, Outcome oder Endpunkt. Wir können die Sprungweite eben sehr einfach mit einer Kommazahl messen. Dafür benötigen wir nur ein Lineal. Das geht schlecht, wenn wir messen, ob einer unserer Hundeflöhe nieste und somit Flohschnupfen hat. Ein Lineal hilft uns hier nicht weiter. Was ähnliches haben wir auch bei der Anzahl der Flohhaare auf dem rechten Flohbein vorliegen. Hier zählen wir und können somit keine halben Flohhaare messen.
Wir sagen, \(Y\) folgt einer Verteilung. Die Sprungweite eines Hundeflohs ist normalverteilt, die Anzahl an Flöhen folgt einer Poisson Verteilung, die Boniturnoten sind multinominal/ordinal bzw. kategoriell verteilt. Der Infektionsstatus ist binomial verteilt. Wir werden uns später die Verteilungen anschauen und visualisieren. Das können wir hier aber noch nicht. Wichtig ist, dass du schon mal gehört hast, dass \(Y\) unterschiedlich verteilt ist, je nachdem welche Dinge wir messen. Die Tabelle 5.1 zeigt dir die Darstellung der Daten von oben in einer einzigen Tabelle. Bitte beachte, dass genau eine Zeile für eine Beobachtung, in diesem Fall einem Floh, vorgesehen ist.
animal gibt an, dass wir es hier mit Hundeflöhe zu tun haben. Die Tabelle ist im Long-Format dargestellt.
| animal | jump_length | flea_count | weight | grade | infected |
|---|---|---|---|---|---|
| dog | 5.7 | 18 | 2.1 | 8 | 0 |
| dog | 8.9 | 22 | 2.3 | 8 | 1 |
| dog | 11.8 | 17 | 2.8 | 6 | 1 |
| dog | 5.6 | 12 | 2.4 | 8 | 0 |
| dog | 9.1 | 23 | 1.2 | 7 | 1 |
| dog | 8.2 | 18 | 4.1 | 7 | 0 |
| dog | 7.6 | 21 | 3.2 | 9 | 0 |
Du findest die Datei flea_dog.xlsx auf GitHub jkruppa.github.io/data/ als Excel oder auch als CSV.
Wir wollen jetzt das Beispiel von den Hundeflöhen um eine weitere Spezies erweitern. Wir nehmen noch die Katzen mit dazu und fragen uns, wie sieht es mit der Sprungfähigkeit von Katzenflöhen aus? Konzentrieren wir uns hier einmal auf die Sprungweite. Wir können wie in dem vorherigen Beispiel mit den Hundeflöhen die Sprungweiten [cm] der Katzenflöhe wieder in der gleichen Weise aufschreiben:
\[ Y_{jump} = \{3.2, 2.2, 5.4, 4.1, 4.3, 7.9, 6.1\}. \]
Wenn wir jetzt die Sprungweiten der Hundeflöhe mit den Katzenflöhen vergleichen wollen haben wir ein Problem. Beide Zahlenvektoren heißen gleich, nämlich \(Y_{jump}\). Wir könnten jeweils in die Indizes noch \(dog\) und \(cat\) schreiben als \(Y_{jump,\, dog}\) und \(Y_{jump,\, cat}\) und erhalten folgende Vektoren. In der Mathematik würden wir jetzt Indizes vergeben, aber das macht die Sachlage nur begrenzt übersichtlicher.
\[ \begin{align} Y_{jump,\, dog} &= \{5.7, 8.9, 11.8, 5.6, 9.1, 8.2, 7.6\}\\ Y_{jump,\, cat} &= \{3.2, 2.2, 5.4, 4.1, 4.3, 7.9, 6.1\} \end{align} \]
Durch mehr Spezies werden die Indizes immer länger und unübersichtlicher. Auch das \(Y\) einfach \(Y_{dog}\) oder \(Y_{cat}\) zu nennen ist keine Lösung - wir wollen uns vielleicht später nicht nur die Sprungweite zwischen den Hunde- und Katzenflöhen vergleichen, sondern vielleicht auch die Anzahl an Flohhaaren oder den Infektionsstatus. Dann stünden wir wieder vor dem Problem die \(Y\) für die verschiedenen Outcomes zu unterscheiden. Daher erstellen wir uns die Tabelle 5.2. Wir haben jetzt eine Datentabelle vorliegen in der die Sprungweiten von sieben Hunden und sieben Katzen dargestellt sind.
| dog | cat |
|---|---|
| 5.7 | 3.2 |
| 8.9 | 2.2 |
| 11.8 | 5.4 |
| 5.6 | 4.1 |
| 9.1 | 4.3 |
| 8.2 | 7.9 |
| 7.6 | 6.1 |
Intuitiv ist die obige Tabelle 5.2 übersichtlich und beinhaltet die Informationen die wir wollten. Wir würden sowas auch schnell in Excel bauen. Für sehr kleine Tabellen ist das auch okay, aber wir werden uns später sehr schnell mit komplexeren Fragestellungen beschäftigen und dann funktioniert das alles nicht mehr. Wir haben nämlich das Problem, das wir in dieser Tabelle 5.2 nicht noch weitere Outcomes angeben können. Wir könnten die Anzahl an Flohhaaren auf den Hunde- und Katzenflöhen nicht einfach so in dieser Form darstellen. Als Lösung ändern wir die Tabelle 5.2 in das Long-Format und erhalten die folgende Tabelle 5.3. Jede Beobachtung belegt nun eine Zeile. Dies ist sehr wichtig im Kopf zu behalten, wenn du eigene Daten in z.B. Excel erstellst.
animal gibt an, dass wir es hier mit Hunde- und Katzenflöhe zu tun haben. Die Tabelle ist im Long-Format dargestellt.
| animal | jump_length | flea_count | weight | grade | infected |
|---|---|---|---|---|---|
| dog | 5.7 | 18 | 2.1 | 8 | 0 |
| dog | 8.9 | 22 | 2.3 | 8 | 1 |
| dog | 11.8 | 17 | 2.8 | 6 | 1 |
| dog | 5.6 | 12 | 2.4 | 8 | 0 |
| dog | 9.1 | 23 | 1.2 | 7 | 1 |
| dog | 8.2 | 18 | 4.1 | 7 | 0 |
| dog | 7.6 | 21 | 3.2 | 9 | 0 |
| cat | 3.2 | 12 | 1.1 | 7 | 1 |
| cat | 2.2 | 13 | 2.1 | 5 | 0 |
| cat | 5.4 | 11 | 2.4 | 7 | 0 |
| cat | 4.1 | 12 | 2.1 | 6 | 0 |
| cat | 4.3 | 16 | 1.5 | 6 | 1 |
| cat | 7.9 | 9 | 3.7 | 6 | 0 |
| cat | 6.1 | 7 | 2.9 | 5 | 0 |
Das Datenformat in der obigen Tabelle nennen wir auch tidy (deu. sauber) nach dem R Paket tidyr was dann auch später mit die Basis für unsere Analysen in R sein wird. Wenn ein Datensatz tidy ist, dann erfüllt er folgende Bedingungen.
Nach diesen Regeln bauen wir dann jeden Datensatz auf, den wir in einem Experiment gemessen haben.
Du findest die Datei flea_dog_cat.xlsx auf GitHub jkruppa.github.io/data/ als Excel oder auch als CSV.
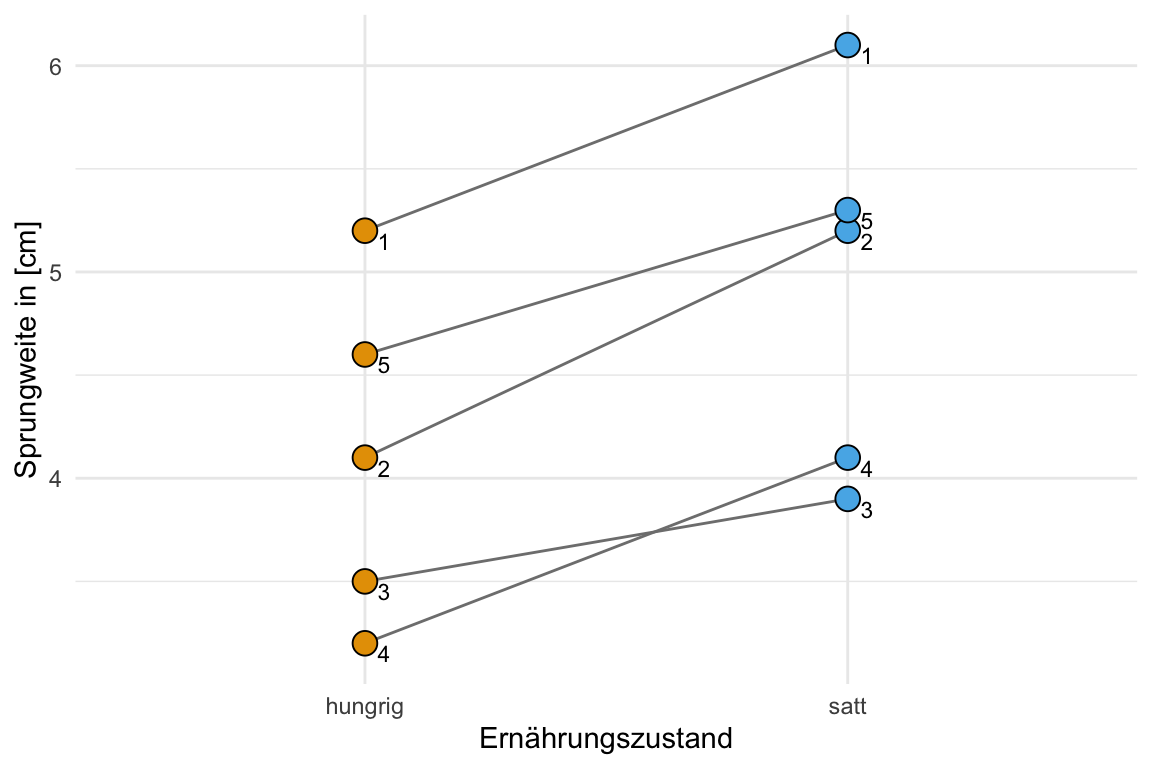
Dann brauchen wir noch einen Datensatz für wiederholte Messungen der Sprungweite in [cm]. Das heitß wir haben nur fünf Hundeflöhe und messen die Sprungweite der Hundeflöhe zu zwei Zeitpunkten. Wir messen einmal wie weit die Flöhe springen würden, wenn die Flöhe hungrig sind. Dann messen wir nochmal die gleichen Flöhe, wenn sich die Flöhe satt gegessen haben. Wir erhalten damit folgende Tabelle.
| id | hungrig | satt |
|---|---|---|
| 1 | 5.2 | 6.1 |
| 2 | 4.1 | 5.2 |
| 3 | 3.5 | 3.9 |
| 4 | 3.2 | 4.1 |
| 5 | 4.6 | 5.3 |
Manchmal sind die Daten schwer zu verstehen, wenn wir nur die Datentabelle vorliegen haben. Deshalb habe ich dir in der folgenden Abbildung nochmal die Daten als Dotplot mit den verbundenen Messungen an den Flöhen dargestellt. Wie du siehst messen wir jeden Floh zweimal.
Wir wollen jetzt das Beispiel von den Hunde- und Katzenflöhen um eine weitere Spezies erweitern. Warum machen wir das? Später wollen wir uns anschauen, wie sich verschiedene Gruppen oder Behandlungen voneinander unterscheiden. Wir brauchen also mehr Spezies. Wir nehmen noch die Füchse mit dazu und fragen uns, wie sieht es mit der Sprungfähigkeit und den anderen Messwerten von Hunde-, Katzen- und Fuchsflöhen aus?
animal gibt an, welche Flohspezies gemessen wurde. Die Tabelle ist im Long-Format dargestellt.
| animal | jump_length | flea_count | weight | grade | infected |
|---|---|---|---|---|---|
| dog | 5.7 | 18 | 2.1 | 8 | 0 |
| dog | 8.9 | 22 | 2.3 | 8 | 1 |
| dog | 11.8 | 17 | 2.8 | 6 | 1 |
| dog | 5.6 | 12 | 2.4 | 8 | 0 |
| dog | 9.1 | 23 | 1.2 | 7 | 1 |
| dog | 8.2 | 18 | 4.1 | 7 | 0 |
| dog | 7.6 | 21 | 3.2 | 9 | 0 |
| cat | 3.2 | 12 | 1.1 | 7 | 1 |
| cat | 2.2 | 13 | 2.1 | 5 | 0 |
| cat | 5.4 | 11 | 2.4 | 7 | 0 |
| cat | 4.1 | 12 | 2.1 | 6 | 0 |
| cat | 4.3 | 16 | 1.5 | 6 | 1 |
| cat | 7.9 | 9 | 3.7 | 6 | 0 |
| cat | 6.1 | 7 | 2.9 | 5 | 0 |
| fox | 7.7 | 21 | 3.1 | 5 | 1 |
| fox | 8.1 | 25 | 4.2 | 4 | 1 |
| fox | 9.1 | 31 | 5.1 | 4 | 1 |
| fox | 9.7 | 12 | 3.5 | 5 | 1 |
| fox | 10.6 | 28 | 3.2 | 4 | 0 |
| fox | 8.6 | 18 | 4.6 | 4 | 1 |
| fox | 10.3 | 19 | 3.7 | 3 | 0 |
Der Datensatz in Tabelle 5.5 beginnt schon recht groß zu werden. Deshalb brauchen wir auch die Statistiksoftware R als Werkzeug um große Datensätze auswerten zu können.
Du findest die Datei flea_dog_cat_fox.xlsx auf GitHub jkruppa.github.io/data/ als Excel oder auch als CSV.
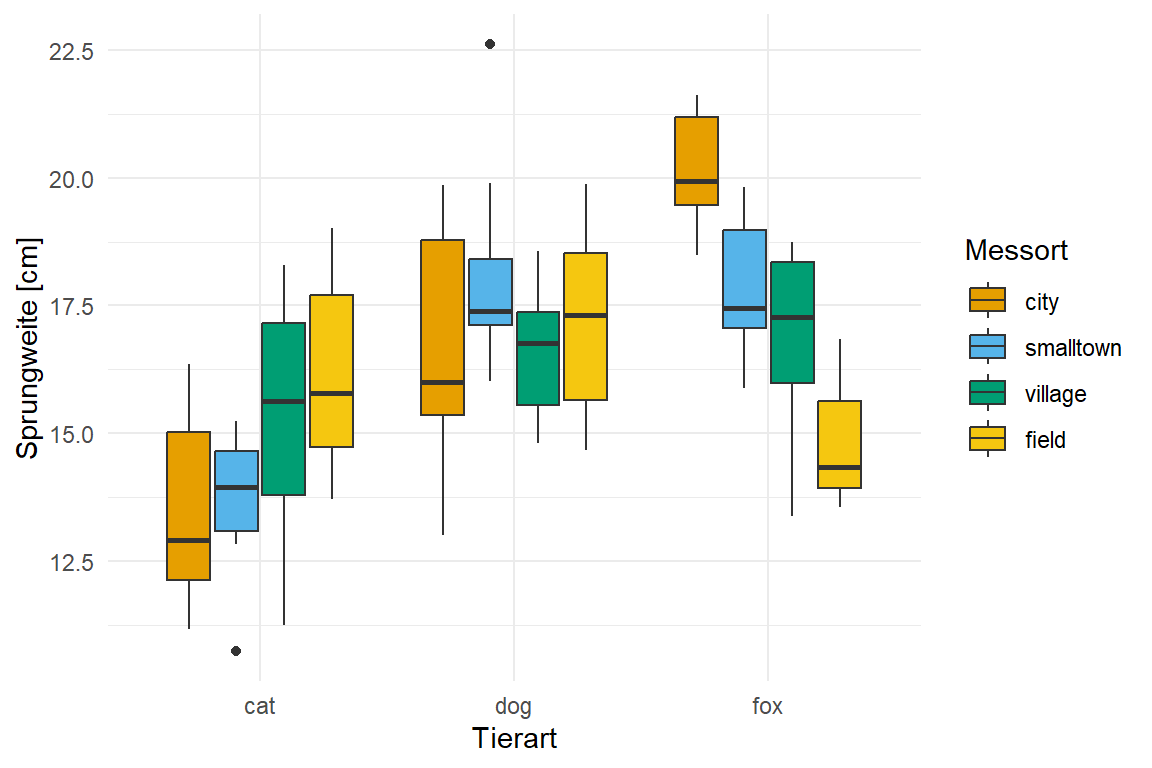
Wir schauen uns in diesem Beispiel wiederum drei Tierarten an: Hunde, Katzen und Füchse. Auf diesen Tierarten messen wir die Sprunglänge von jeweils zehn Tieren, lassen aber den Rest einmal weg. Im Vergleich zu dem vorherigen Beispiel erweitern wir die Daten um eine Spalte site in der wir vier verschiedene Messorte protokollieren. Es ergibt sich folgende Tabelle 5.6 und die dazugehörige Abbildung 5.3.
| animal | site | rep | jump_length |
|---|---|---|---|
| cat | city | 1 | 12.04 |
| cat | city | 2 | 11.98 |
| cat | city | 3 | 16.1 |
| cat | city | 4 | 13.42 |
| cat | city | 5 | 12.37 |
| cat | city | 6 | 16.36 |
| … | … | … | … |
| fox | field | 5 | 16.38 |
| fox | field | 6 | 14.59 |
| fox | field | 7 | 14.03 |
| fox | field | 8 | 13.63 |
| fox | field | 9 | 14.09 |
| fox | field | 10 | 15.52 |
Die Datentabelle ist in dieser Form schon fast nicht mehr überschaubar. Daher hilft hier die explorative Datenanalyse weiter. Wir schauen uns daher die Daten einmal als einen Boxplot in Abbildung 5.3 an. Wir sehen hier, dass wir drei Tierarten an vier Orten die Sprungweite in [cm] gemessen haben.
Du findest die Datei flea_dog_cat_fox_site.xlsx auf GitHub jkruppa.github.io/data/ als Excel oder auch als CSV.
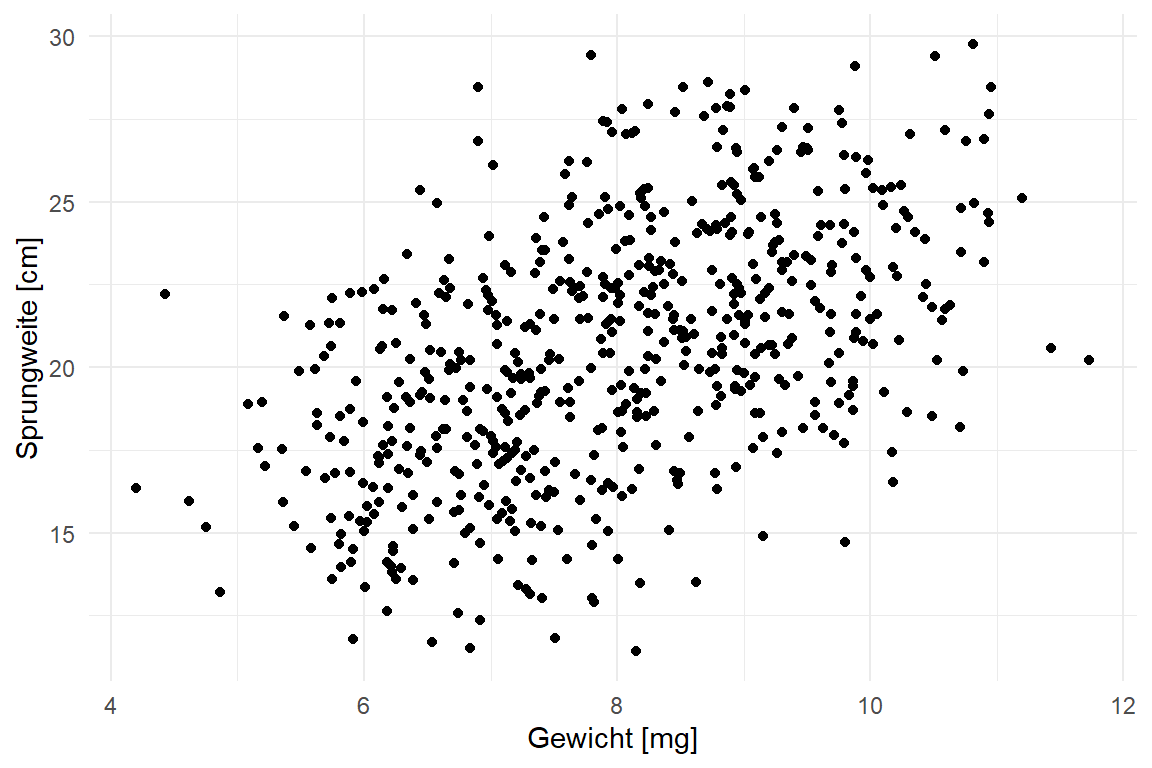
Wir schauen uns in diesem Beispiel wiederum nur zwei Tierarten an: Hunde und Katzen. Auf diesen Tierarten messen wir wieder die Sprunglänge in [cm] von jeweils 400 Tieren. Im Vergleich zu dem vorherigen Beispiel erweitern wir die Daten um eine Spalte jump_weight in [mg] sowie sex [male, female]. Bei Versuch wurde noch in der Variable hatch_time gemessen, wie lange die Flöhe in Stunden zum Schlüpfen brauchen. Es ergibt sich folgende Tabelle 5.7 mit den ersten zehn Beobachtungen und die dazugehörige Abbildung 5.4.
| animal | sex | weight | jump_length | flea_count | hatch_time |
|---|---|---|---|---|---|
| cat | male | 6.02 | 15.79 | 5 | 483.60 |
| cat | male | 5.99 | 18.33 | 1 | 82.56 |
| cat | male | 8.05 | 17.58 | 1 | 296.73 |
| cat | male | 6.71 | 14.09 | 3 | 140.90 |
| cat | male | 6.19 | 18.22 | 1 | 162.20 |
| cat | male | 8.18 | 13.49 | 1 | 167.47 |
| cat | male | 7.46 | 16.28 | 1 | 291.20 |
| cat | male | 5.58 | 14.54 | 0 | 112.58 |
| cat | male | 6.19 | 16.36 | 1 | 143.97 |
| cat | male | 7.53 | 15.08 | 1 | 766.31 |
Die Datentabelle ist in dieser Form schon fast nicht mehr überschaubar. Daher hilft hier die explorative Datenanalyse weiter. Wir schauen uns daher die Daten einmal als einen Scatterplot in Abbildung 5.4 an. Wir sehen hier, dass wir das mit dem Gewicht [mg] der Flöhe auch die Sprungweite in [cm] steigt.
Du findest die Datei flea_dog_cat_length_weight.xlsx auf GitHub jkruppa.github.io/data/ als Excel oder auch als CSV.

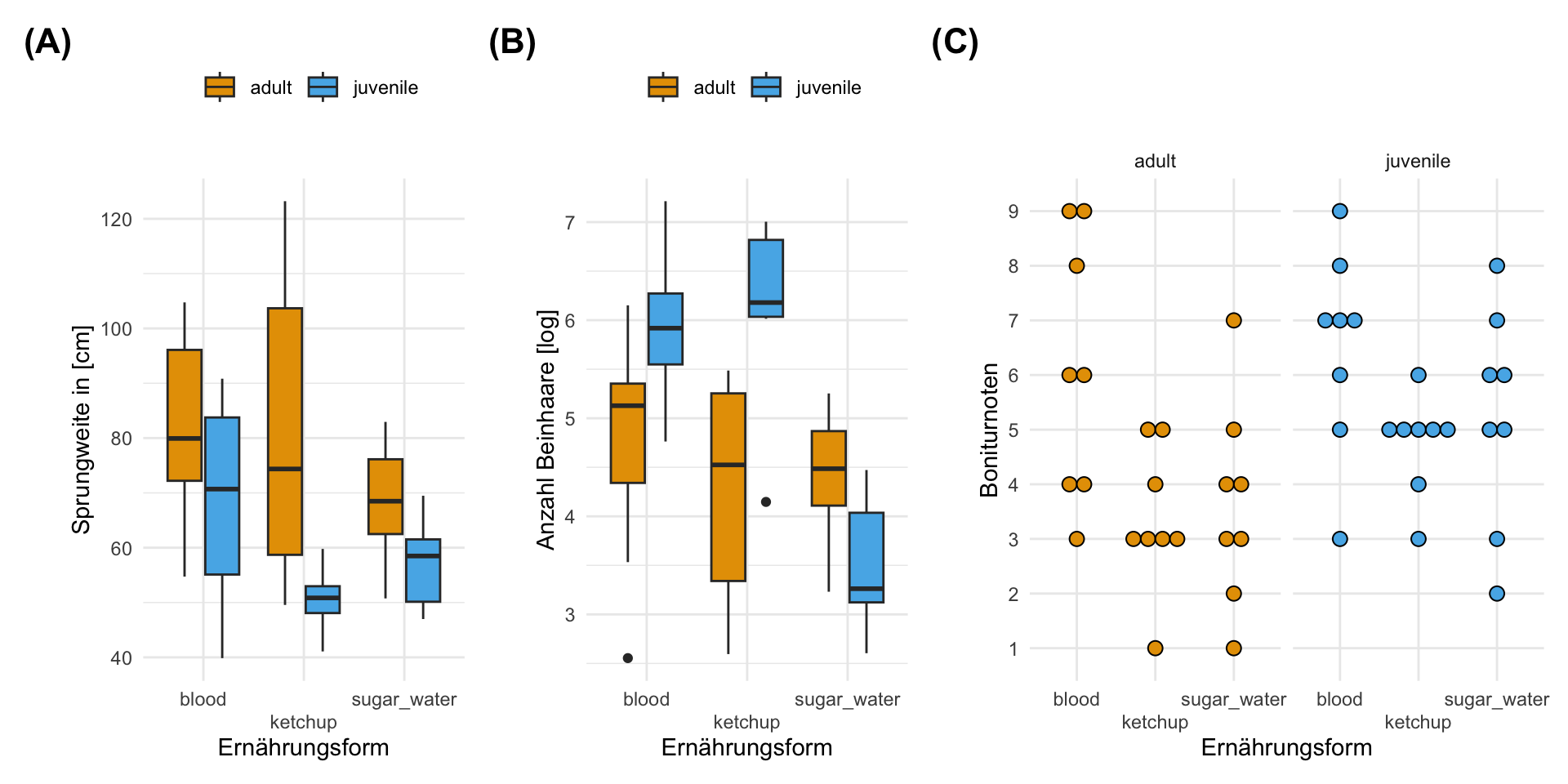
Wenn wir uns später komplexere Modelle anschauen, dann brauchen wir auch etwas komplexere Daten. Im Folgenden habe ich einmal die Flohdaten erweitert. Wir fragen uns hier, ob die Fütterung der Flöhe einen Unterschied auf die Sprungweite hat. Darüber hinaus schauen wir uns auch noch verschiedene Entwicklungsstadien der Flöhe an. Daher haben wir hier ein zweifaktorielles Design vorliegen. Zusätzlich haben wir noch das Gewicht der Flöhe bestimmt und mit wie vielen Flöhen der einzelne Floh geschlüpft ist. Wir haben also folgende Faktoren und Einflussfaktoren vorliegen.
feeding mit den Leveln Blut blood mit [50g/ml], Zuckerwasser als Kontrolle sugar_water mit [25g/ml] und Ketchup ketchup mit [1ml] pro Fütterung.stage mit den zwei Leveln für juvenile Flöhe juvenile und ausgewachsenen Flöhen adult.weight in [mg] der einzelnen Flöhe zum Zeitpunkt der Messung.hatched des einzelnen Flohes. Damit auch die Anzahl an Geschwistern in dem jeweiligen Wurf aus dem der Floh stammt.Im Weiteren haben wir unterschiedliche Outcomes \(y\) gemessen. Zum einen die Sprungweite in [cm], die Exterieurbeurteilung der Flöhe als Bonitur, die Anzahl an Haaren an dem linken sowie rechten Bein sowie der Infektionsstatus der Flöhe mit Flohschnupfen. Alle vier Messwerte folgen damit dann auch einer anderen Verteilung und haben somit auch einen anderen Zahlenraum.
jump_length gemessen in [cm] gesprungen.count_leg_left und count_leg_right sowie die mittlere, aufgerundete Anzahl mit count_leg, als ein weiterer Indikator für Gesundheit und Aerodynamik.bonitur auf einer Likert Skala von 1 bis 9, wobei 9 die stärkste Ausprägung und damit den gesundesten Floh darstellt.infected auf einer Skala von infiziert \((ja/1)\) und nicht infiziert \((nein/0)\).Es ergibt sich dann die folgende Abbildung der drei Endpunkte für die Sprungweite, gemittelte Anzahl an Haaren an den Beinen auf der log-Skala sowie die Exterieurbeurteilung. Der binäre Endpunkt mit dem Infektionsstatus lässt sich immer schwerer in einer Abbilung darstellen. Daher ergänze ich dazu noch die Tabelle 54.9 für eine bessere Übersicht.
Die folgende Tabelle zeigt nochmal die Verteilung der gesunden und kranken Flöhe mit Flohschnupfen aufgeteilt für die Faktoren Ernährungsform sowie Entwicklungsstand. Dazu kommt dann noch das mittlere Gewicht in den beiden Infektionsgruppen sowie die mittlere Anzahl an Geschwistern in dem entsprechenden Wurf. Wir haben eine ungefähre Gleichverteilung von gesunden zu kranken Flöhen.
| Characteristic | 0 N = 211 |
1 N = 271 |
|---|---|---|
| feeding | ||
| blood | 3 / 21 (14%) | 13 / 27 (48%) |
| ketchup | 9 / 21 (43%) | 7 / 27 (26%) |
| sugar_water | 9 / 21 (43%) | 7 / 27 (26%) |
| stage | ||
| adult | 14 / 21 (67%) | 10 / 27 (37%) |
| juvenile | 7 / 21 (33%) | 17 / 27 (63%) |
| weight | 11.18 (5.88) | 9.00 (5.05) |
| hatched | 478.63 (249.99) | 455.82 (194.37) |
| 1 n / N (%); Mean (SD) | ||
Nach dem Ende der Laufzeit unseres Experiments haben wir dann auch noch jedem Floh einmal Blut abgenommen und geschaut wie die Laborwerte aussehen. Dazu haben wir dann aber natürlich nur eine Auswahl an sechs Laborwerten genommen. Wir haben folgende sechs Werte aus dem Blut der Flöhe bestimmt.
Es ergeben sich dann folgende Boxplots in der Abbildung 5.7. Auf den ersten Blick sehen die kontinuierlichen Laborwerte alle sehr unterschiedlich aus. Den noch magst du einwenden, dass eventuell die Laborwerte doch irgendwie zusammenhängen könnten. So könnte man annehmen, dass eventuell auch hohe Kaliumwerte mit hohen Magnesiumwerten einhergehen. Solche Korrelationen werden wir uns dann in den folgenden Kapiteln anschauen.
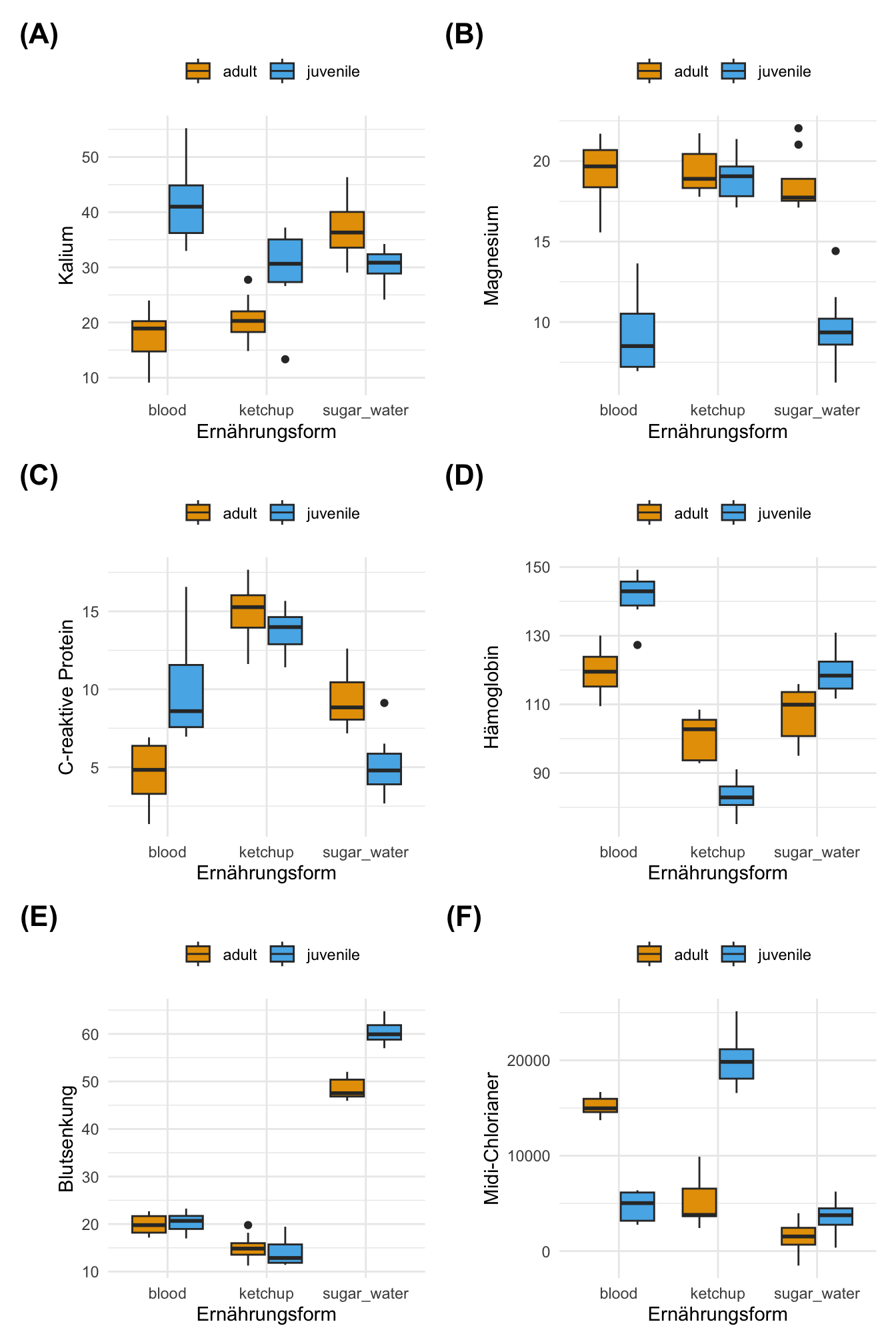
Dann haben wir einmal einen komplexen Datensatz, den wir uns dann detaillierter anschauen können. Du findest dann den Datensatz auch auf GitHub, wie du im folgenden Kasten siehst.
Du findest die Datei fleas_model_data.xlsx auf GitHub jkruppa.github.io/data/ als Excel.