```{r echo = FALSE}
pacman::p_load(tidyverse, readxl, knitr, kableExtra)
set.seed(20230526)
```
# Linear Probability Model {#sec-prob-model}
*Letzte Änderung am `r format(fs::file_info("stat-modeling-prob-model.qmd")$modification_time, '%d. %B %Y um %H:%M:%S')`*
Das Wahrscheinlichkeitsmodell (eng. *probability model* oder *linear probability model*) ist ein seltsames Konstrukt aus der Ökonomie und den Sozialwissenschaften. Wir nutzen hier auch den englischen Begriff, da der deutsche Begriff eigentlich nicht benutzt wird. Es handelt sich hier also um *probability models*. Was macht also das *probability model*? Eigentlich alles falsch, was man so im Allgemeinen meinen würde. Das *probability model* nimmt als Outcome $y$ eine $0/1$ Variable. Wir modellieren also wie bei der logistischen Regression ein Outcome $y$ was sich nur durch zwei Ausprägungen darstellen lässt. Und damit wären wir dann auch beim Punkt angekommen. Anstatt das $0/1$ Outcome jetzt *richtig* mit der logistischen Regression auszuwerten, nutzen wir die klassische Gaussian Regression mit der Annahme eines normalverteilten Outcomes. Wir nutzen hier also das falsche Modell für das gemessene Outcome. Warum machen wir das? Weil sich die Effektschätzer aus einer Gaussian Regression auf einem $0/1$ Outcome besser interpretieren lassen. Wie immer gibt es viel Disput, ob du so was überhaupt machen darfst. Können tust es auf jeden Fall. Wir können auf alle Daten alle Modelle anwenden. Nun ist es aber so, dass die Auswertung von einem $0/1$ Outcome mit einem *probability model* teilweise in der Ökonomie oder den Sozialwissenschaften sehr verbreitet ist. Deshalb findest du auch hier dieses etwas kurzes Kapitel.
Wir immer gibt es auch wieder zwei gute Tutorien auf die sich hier alles reimt. Einmal bitte das Tutorium [Linear Probability Model](https://murraylax.org/rtutorials/linearprob.html) sowie [Binary Dependent Variables and the Linear Probability Model](https://www.econometrics-with-r.org/11-1-binary-dependent-variables-and-the-linear-probability-model.html) besuchen. Wenn du noch mehr über `lm()` und `glm()` lesen möchtest, dann kannst du das auch nochmal in der Frage zu [Linear probability model: lm() und glm()](https://stats.stackexchange.com/questions/489651/linear-probability-model-why-do-lm-and-glm-not-give-the-same-results-in-r) machen.
## Genutzte R Pakete
Wir wollen folgende R Pakete in diesem Kapitel nutzen.
```{r echo = TRUE}
#| message: false
pacman::p_load(tidyverse, magrittr, sandwich, lmtest,
emmeans, multcomp, see,
performance, broom, conflicted)
conflicts_prefer(dplyr::select)
conflicts_prefer(magrittr::extract)
conflicts_prefer(magrittr::set_names)
```
An der Seite des Kapitels findest du den Link *Quellcode anzeigen*, über den du Zugang zum gesamten R-Code dieses Kapitels erhältst.
## Daten
In diesem Kapitel nutzen wir die infizierten Ferkel als Beispieldatensatz. Wir haben in dem Datensatz über vierhundert Ferkel untersucht und festgehalten, ob die Ferkel infiziert sind ($1$, ja) oder nicht infiziert ($0$, nein). Wir haben daneben noch eine ganze Reihe von *Risiko*faktoren erhoben. Hier sieht man mal wieder wie wirr die Sprache der Statistik ist. Weil wir rausfinden wollen welche Variable das Risiko für die Infektion erhöht, nennen wir diese Variablen Risikofaktoren. Wir nehmen hier jetzt aber nicht alle Variablen mit, sondern nur die Variablen für den Entzündungswert `crp`, das Geschlecht `sex`, dem Alter `age` und der Gebrechlichkeitskategorie `frailty`.
```{r}
#| message: false
pig_tbl <- read_excel("data/infected_pigs.xlsx") |>
select(infected, crp, sex, age, frailty)
```
Schauen wir uns nochmal einen Ausschnitt der Daten in der @tbl-lpm-pigs an.
```{r}
#| echo: false
#| message: false
#| warning: false
#| label: tbl-lpm-pigs
#| tbl-cap: Auszug aus dem Daten zu den kranken Ferkeln.
rbind(head(pig_tbl, 4),
rep("...", times = ncol(pig_tbl)),
tail(pig_tbl, 4)) |>
kable(align = "c", "pipe")
```
Im Folgenden wollen wir einmal modellieren, ob es einen Zusammenhang von den Variablen `crp`, `sex`, `age` und `frailty` auf das $0/1$-Outcome `infected` gibt. Welche der Variablen erniedrigen oder erhöhen also das Risiko einer Ferkelinfektion?
## Theoretischer Hintergrund
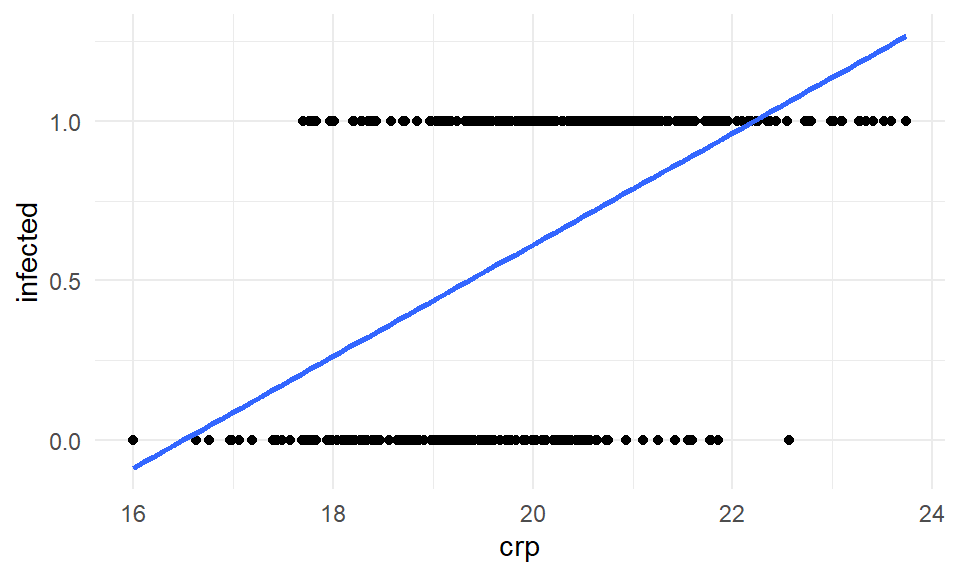
Den theoretischen Hintergrund belassen wir hier nur kurz. Die Idee sehen wir dann einmal in der @fig-lpm-activity-01. Wir haben hier dann nur die Variable `crp` und das Outcome `infected` dargestellt. Wir sehe die klaren zwei Ebenen. Wir haben ja bei dem Infektionsstatus auch nur zwei mögliche Ausprägungen. Entweder ist unser Ferkel infiziert oder eben nicht. Da wir aber die Entzündungswerte kontinuierlich messen ergeben sich die beiden Ebenen.
```{r}
#| echo: true
#| message: false
#| label: fig-lpm-activity-01
#| fig-align: center
#| fig-height: 3
#| fig-width: 5
#| fig-cap: "Visualisierung des Zusammenhangs zwischen dem Infektionsstatus und den Entzündungswerten der Ferkel."
ggplot(pig_tbl, aes(x = crp, y = infected)) +
theme_minimal() +
geom_point()
```
Tja und dann rechnen wir einfach eine Gaussian linear Regression mit der Funktion `lm()`. Es ergibt sich dann eine gerade Linie, wie wir sie in der @fig-lpm-activity-02 sehen. Da wir aber mit unserem Infektionsstatus auf $0/1$ begrenzt sind, aber eine Gerade nicht, haben wir das Problem, dass wir in diesem Fall den Infektionsstatus für CRP-Werte größer als 22 überschätzen. Ich meine mit überschätzen dann auch wirklich Werte zu erhalten, die es dann gar nicht geben kann. Es kann keinen Infektionsstatus über *ja* geben.
```{r}
#| echo: true
#| message: false
#| label: fig-lpm-activity-02
#| fig-align: center
#| fig-height: 3
#| fig-width: 5
#| fig-cap: "Visualisierung des Zusammenhangs zwischen dem Infektionsstatus und den Entzündungswerten der Ferkel ergänzt um Gerade aus der Gaussian linearen Regression."
ggplot(pig_tbl, aes(x = crp, y = infected)) +
theme_minimal() +
geom_point() +
stat_smooth(method = "lm", se = FALSE)
```
## Modellierung
Dann können wir schon das *probability model* anpassen. Dazu nehmen wir die Funktion `lm()`, die wir auch für unsere Gaussian linearen Regression unter der Annahme eines normalverteilten Outcomes $y$ nutzen. Wichtig ist hier, dass wir auf keinen Fall unseren Infektionsstatus `infected` als einen Faktor übergeben. Der Infektionsstatus `infected` muss numerisch sein.
```{r}
lm_fit <- lm(infected ~ crp + age + sex + frailty, data = pig_tbl)
```
Schauen wir uns aber gleich mal die Modellausgabe an. Wie immer, du kannst alle Zahlen und Spalten in eine Funktion stecken und am Ende kommt dann was raus. Woher soll auch die Funktion wissen, dass es sich um einen Faktor handelt oder eine numerische Variable?
```{r}
lm_fit |>
summary()
```
Neben der sehr schlechten Modellgüte, die wir am Bestimmtheitsmaß $R^2$ mit 0.24 erkennen, sind aber die Residuen nach den deskriptiven Maßzahlen einigermaßen okay. Wir werden aber gleich noch sehen, dass die Maßzahlen hier auch trügen können. Was ist den nun das Tolle am *probability model*? Wir können die Effektschätzer `Estimate` direkt als prozentuale Veränderung interpretieren. Das heißt, wir können sagen, dass pro Einheit `crp` die Wahrscheinlichkeit infiziert zu sein um 16.2087% ansteigt. Das ist natürlich eine sehr schöne Eigenschaft. Nur leider gibt es da meistens dann doch ein Problem. Dafür schauen wir uns einmal die Spannweite der vorhergesagten Werte an.
```{r}
lm_fit |>
predict() |>
range() |>
round(2)
```
Wie du siehst, kriegen wir Werte größer als Eins und kleiner als Null aus dem Modell raus. Das macht jetzt aber recht wenig Sinn. Wir können die Werte aus `predict()` als Wahrscheinlichkeit infiziert zu sein interpretieren. Da unsere Ferkel aber nur gesund oder krank sein können, machen negative Werte der Wahrscheinlichkeit infiziert zu sein keinen Sinn. Auch Werte größer als Eins können wir nur sehr schwer interpretieren.
## Varianzheterogenität
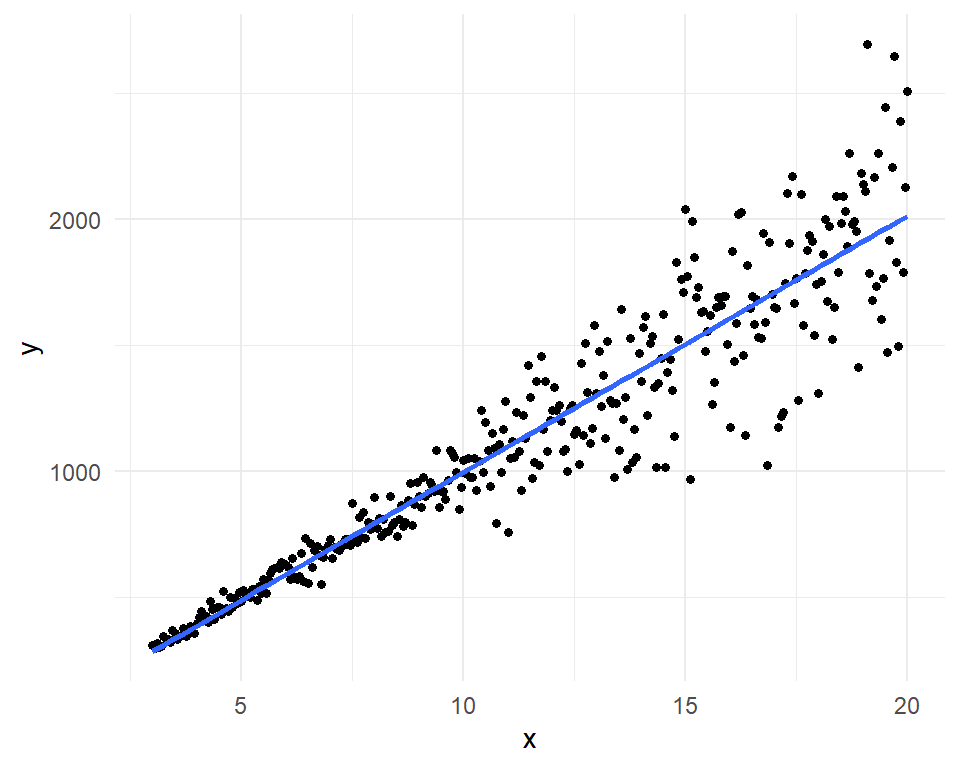
Wenn du das *probability model* durchführst, dann hast du in den meisten Fällen das Problem der Heteroskedastizität, auch Varianzheterogenität genannt. Oder in anderen Worten, die Residuen als Fehler unseres Modell sind nicht gleichmäßig mit gleich großen Werten verteilt um die Gerade. Gut, dass klingt jetzt etwas sperrig, hier einmal die @fig-lpm-var-hetero um es besser zu verstehen.
```{r}
#| echo: false
#| message: false
#| label: fig-lpm-var-hetero
#| fig-align: center
#| fig-height: 4
#| fig-width: 5
#| fig-cap: "Varianzheterogenität oder Heteroskedastizität in den Daten. Die Abstände der Punkte zu der Geraden, die Residuen, werden immer größer. Wir haben keinen konstanten oder homogenen Fehler."
var_hetro_tbl <- tibble(x = seq(3, 20, by = 0.05),
y = 100 * x + rnorm(length(x), 0, x^2))
ggplot(var_hetro_tbl, aes(x, y)) +
theme_minimal() +
geom_point2() +
stat_smooth(method = "lm", se = FALSE)
```
Für unser Modell `lm_fit` von oben können wir auch gleich die Funktion `check_heteroscedasticity()` aus dem R Paket `{performance}` nutzen um zu Überprüfen ob wir Varianzheterogenität vorliegen haben. Aber Achtung, ich wäre hier sehr vorsichtig, wenn die Funktion sagt, dass wir keine Varianzheterogenität vorliegen haben.
```{r}
check_heteroscedasticity(lm_fit)
```
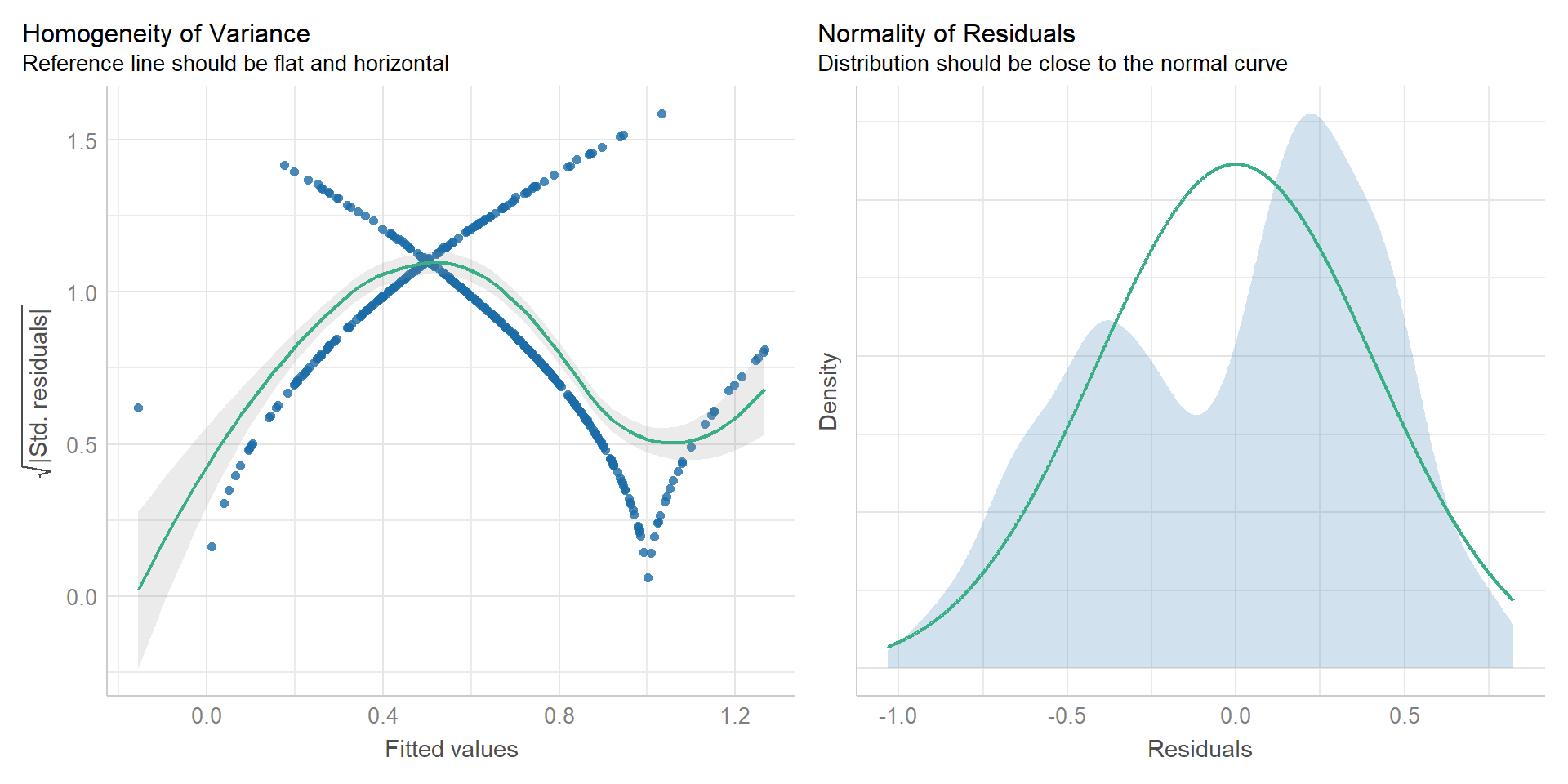
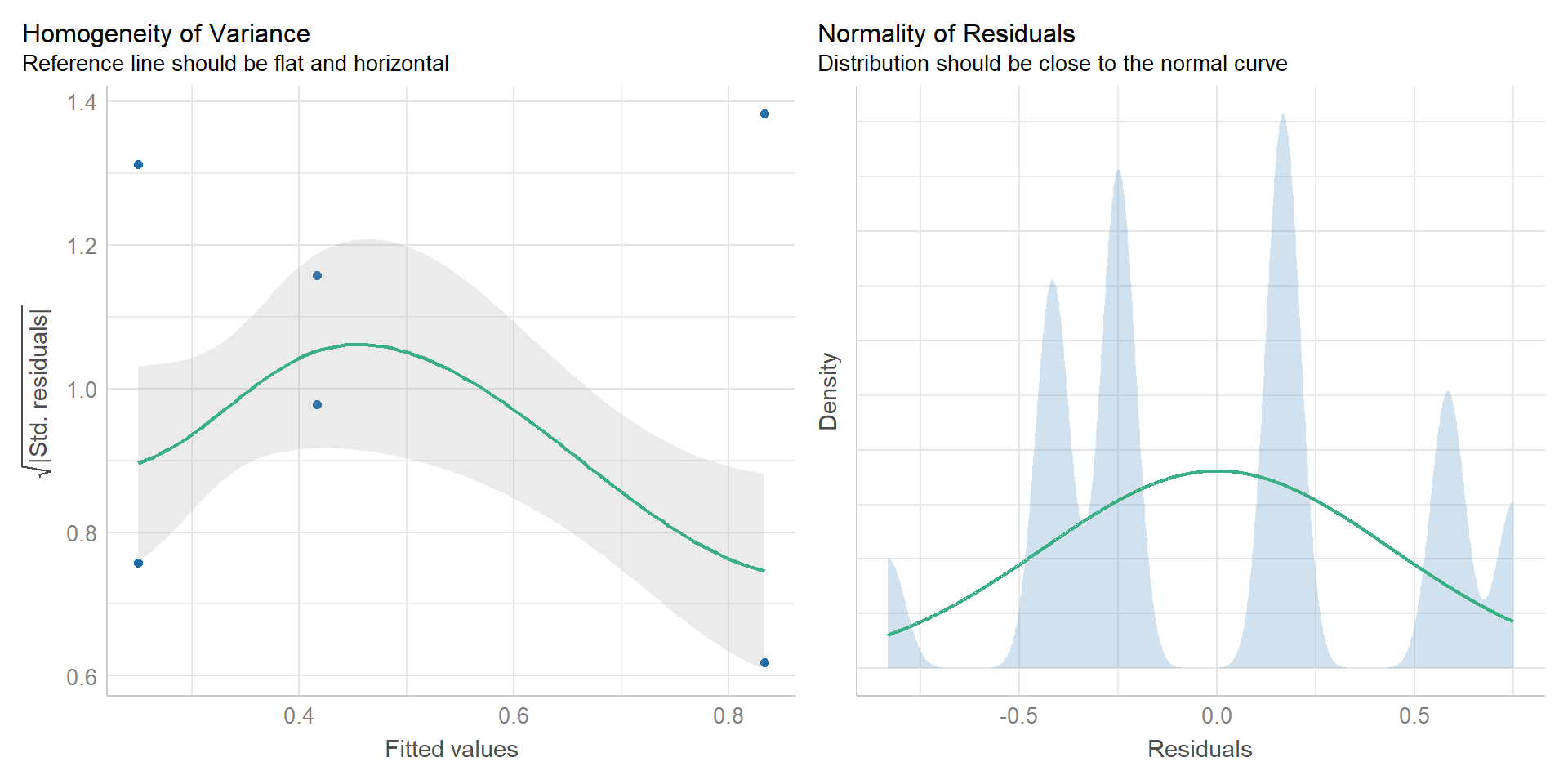
Neben der Funktion `check_heteroscedasticity()` gibt es auch die Möglichkeit über `check_model()` sich die Varianzen und damit die Residuen einmal anzuschauen. Die visuelle Überprüfung ist auf jeden Fall Pflicht. Und wie du in der @fig-lpm-02 siehst, sind die Varianzen weder homogen noch irgendwie normalverteilt. Wir gehen also von Varianzheterogenität aus. Damit liegen wir in Linie mit der Funktion `check_heteroscedasticity()`, aber das muss nicht immer unbedingt sein. Besonders bei kleiner Fallzahl, kann es vorkommen, dass `check_heteroscedasticity()` eine Varianzheterogenität übersieht.
```{r}
#| echo: true
#| message: false
#| label: fig-lpm-02
#| fig-align: center
#| fig-height: 5
#| fig-width: 10
#| fig-cap: "Überprüfung der Varianzhomogeniutät des Modells `lm_fit` mit der Funktion `check_model()`. Wir sehen hier eine klare Varianzheterogenität in dem Modell."
check_model(lm_fit, check = c("homogeneity", "normality"))
```
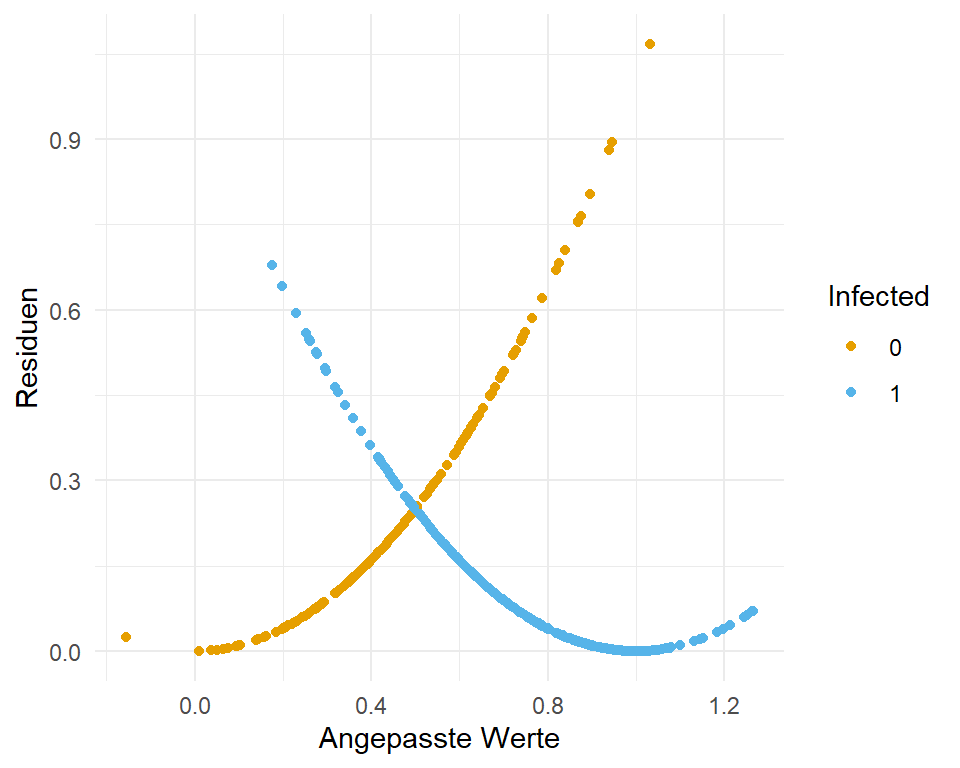
Jetzt kann man sich fragen, warum sind denn da so Bögen drin? Das kommt von den Abständen der Punkte auf den beiden Ebenen. Die Gerade läuft ja durch einen Bereich in dem keine Beobachtungen sind. Daher ist am Anfang der Abstand zu einer der beiden Ebenen, entweder der Null oder der Eins, minimal und erhöht sich dann langsam. Weil ja nicht alle Beobachtungen alle bei Null sind springen die Abstände von klein zu groß. In @fig-lpm-01a siehst du nochmal den Zusammenhang. Unsere angepasste Gerade steigt ja an, wie du in @fig-lpm-activity-02 siehst. Daher sind die Abstände zu den Null Werten am Anfang sehr klein und stiegen dann an. Sobald die erste Beobachtung mit einem Infektionsstatus von Eins auftaucht, springt der Abstand natürlich sofort nach oben. Werte größer als Eins dürfte es auf der x-Achse gar nicht geben, den dort werden dann Werte größer als Eins geschätzt.
```{r}
#| echo: true
#| message: false
#| label: fig-lpm-01a
#| fig-align: center
#| fig-height: 4
#| fig-width: 5
#| fig-cap: "Etwas andere Darstellung der angepassten Werte `.fitted` und der Residuen `.resid` aus dem Modell `lm_fit`. Die Punkte sind nach dem Infektionsstatus eingefärbt."
lm_fit |>
augment() |>
ggplot(aes(x = .fitted, y = .resid^2, color = as_factor(infected))) +
theme_minimal() +
geom_point() +
labs(x = "Angepasste Werte", y = "Residuen", color = "Infected") +
scale_color_okabeito()
```
## Interpretation des Modells
Wir haben ja schon einmal weiter oben in das Modell geschaut und eine Interpretation vorgenommen. Wir erinnern uns, wir können die Effektschätzer `Estimate` aus einem *probability model* direkt als prozentuale Veränderung interpretieren. Das heißt, wir können sagen, dass pro Einheit `crp` die Wahrscheinlichkeit infiziert zu sein um 16.2087% ansteigt. Das ist natürlich eine sehr schöne Eigenschaft. Dann haben wir auch noch gleich die Richtung mit drin, wenn wir also negative Effekte haben, dann senkt die Variable das Risiko pro Einheit um den prozentualen Wert. Bei kategorialen Variablen haben wir dann den Unterschied zu der nicht vorhandenen Gruppe. Daher sind männliche Ferkel um 5% *weniger* infiziert als weibliche Ferkel. Leider geht der t-Test, der die $p$-Werte produziert, von homogenen Varianzen aus. Die haben wir aber nicht vorliegen.
```{r}
lm_fit |>
summary() |>
pluck("coefficients") |>
round(4)
```
Deshalb müssen wir nochmal ran. Wir können die Funktion `coeftest()` aus dem R Paket `{lmtest}` zusammen mit den R Paket `{sandwich}` nutzen um unsere Modellanpassung für die Varianzheterogenität zu adjustieren. Wir ändern also die Spalte `Strd. Error`. Es gibt aber sehr viele Möglichkeiten `type` die Varianz anzupassen. Das ist ein eigenes Kapitel worum wir uns hier nicht scheren. Wir nehmen mehr oder minder den Standard mit `HC1`.
```{r}
lm_fit |>
coeftest(vcov. = vcovHC, type = "HC1") |>
round(4) |>
tidy()
```
Wir schauen also als erstes auf den Standardfehler und sehen, dass unsere Gaussian lineare Regression (OLS) den Standardfehler als zu hoch geschätzt hat. Größer Standardfehler bedeutet kleinere Teststatistik und damit dann auch weniger signifikante $p$-Werte. In der @tbl-lpm-comp siehst du nochmal die beiden Spalten der Standardfehler nebeneinander. Unser Sandwich-Schätzer (HC1) liefert da die besseren Fehlerterme, die eher der Realität der Varianzheterogenität entsprechen. Wir brauchen die adjustierten Standardfehler aber nur, wenn wir eine statistischen Test rechnen wollen und den $p$-Wert für die Bewertung der Signifikanz brauchen.
```{r}
#| echo: false
#| message: false
#| warning: false
#| label: tbl-lpm-comp
#| tbl-cap: "Vergleich der Standardfehler der Gaussian linear Regression mit der Annahme der homogenen Varianzen (OLS) und die Adjusterung der Fehler mit dem Sandwich-Schätzer (HC1)."
bind_cols(names(coef(lm_fit))) |>
bind_cols(lm_fit |>
summary() |>
pluck("coefficients") |>
extract(, 2),
lm_fit |>
coeftest(vcov. = vcovHC, type = "HC1") |>
tidy() |>
pluck("std.error")) |>
mutate_if(is.numeric, round, 4) |>
set_names(c("", "OLS", "HC1")) |>
kable(align = "lcc", "pipe")
```
## Gruppenvergleich
Häufig ist es ja so, dass wir das Modell nur schätzen um dann einen Gruppenvergleich zu rechnen. Das heißt, dass es uns interessiert, ob es einen Unterschied zwischen den Leveln eines Faktors gegeben dem Outcome $y$ gibt. Wir machen den Gruppenvergleich jetzt einmal an der Gebrechlichkeit `frailty` einmal durch. Wir habe die drei Gruppen `frail`, `pre-frail` und `robust` vorliegen. Danach schauen wir uns nochmal die prinzipielle Idee des Gruppenvergleichs auf mittleren Wahrscheinlichkeiten infiziert zu sein an.
Eigentlich ist es recht einfach. Wir nehmen wieder unser lineares Modell, was wir oben schon angepasst haben. Wir schicken dann das Modell in die Funktion `emmeans()` um die Gruppenvergleiche zu rechnen. Jetzt müssen wir nur zwei Dinge noch machen. Zum einen wollen wir alle paarweisen Vergleiche zwischen den drei Leveln von dem Faktor `frailty` rechnen, deshalb setzen wir `method = "pairwise"`. Dann müssen wir noch dafür sorgen, dass wir nicht homogene Varianzen schätzen. Deshalb setzen wir die Option `vcov. = sandwich::vcovHAC`. Damit wählen wir aus dem Paket `{sandwich}` den Sandwichschätzer `vcovHAC` und berücksichtigen damit die Varianzheterogenität in den Daten. Wenn du das Paket `{sandwich}` schon geladen hast, dann musst du das Paket nicht mit Doppelpunkt vor die Funktion des Sandwich-Schätzers setzen.
```{r}
em_obj <- lm_fit |>
emmeans(~ frailty, method = "pairwise", vcov. = sandwich::vcovHAC)
em_obj
```
Dann können wir auch schon uns die Kontraste und damit die Mittelwertsvergleiche wiedergeben lassen. Was heißt hier Mittelwertsvergleiche? Die Mittelwerte sind hier natürlich die mittlere Wahrscheinlichkeit infiziert zu sein. Wir adjustieren hier einmal die $p$-Werte für multiple Vergleiche nach Bonferroni, damit du auch mal die Optionen siehst.
```{r}
em_obj |>
contrast(method = "pairwise", adjust = "bonferroni")
```
Wir sehen also, dass es einen prozentualen Unterschied zwischen `frail - (pre-frail)` von -3% gibt. Daher ist die mittlere Wahrscheinlichkeit von `pre-frail` größer als die von `frail`. Den Zusammenhang sehen wir auch weiter oben in der Ausgabe von `emmeans`. Dort haben wir eine mittlere Infektionswahrscheinlichkeit von 66.8% für `frail` und eine mittlere Infektionswahrscheinlichkeit von 70.6% für `pre-frail`. Keiner der Vergleiche ist signifikant. Beachte, dass jeder Vergleich immer einen unterschiedlichen Standardfehler zugewiesen bekommt um die Varianzheterogenität zu berücksichtigen.
Jetzt lassen wir uns nochmal das unadjustierte *compact letter display* wiedergeben. Aber auch in dem unadjustierten Fall finden wir keine signifikanten Unterschiede.
```{r}
em_obj |>
cld(Letters = letters, adjust = "none")
```
Am Ende möchte ich hier nochmal einen Spieldatensatz `toy_tbl` erstellen indem ich wiederum drei Gruppen miteinander vergleiche. Ich tue mal so als würden wir uns hier zwei Pestizide und eine Kontrolle anschauen. Unser Outcome ist dann, ob wir eine Infektion vorliegen haben oder das Pestizid alles umgebracht hat. Damit haben wir dann unser Outcome `infected` definiert. Wir bauen uns die Daten so, dass 80% der Beobachtungen in der Kontrolle infiziert sind. In den beiden Behandlungsgruppen sind jeweils 50% und 30% der Beobachtungen nach der Behandlung noch infiziert. Wir haben jeweils zwölf Pflanzen `n_grp` beobachtet. Das sind wirklich wenige Beobachtungen für einen $0/1$ Endpunkt.
```{r}
n_grp <- 12
toy_tbl <- tibble(trt = gl(3, n_grp, labels = c("ctrl", "roundUp", "killAll")),
infected = c(rbinom(n_grp, 1, 0.8), rbinom(n_grp, 1, 0.5), rbinom(n_grp, 1, 0.2)))
```
Jetzt bauen wir uns wieder unser Modell zusammen.
```{r}
toy_fit <- lm(infected ~ trt, data = toy_tbl)
toy_fit
```
Wie du sehen kannst, treffen wir die voreingestellten Infektionswahrscheinlichkeiten nur einigermaßen. Wir wollen für die Kontrolle 80% und erhalten 83.3%. Für `roundUp` haben wir 50% gewählt und erhalten $83.3 - 41.67 = 41.63$. Auch bei `killAll` sieht es ähnlich aus, wir wollen 20% und erhalten `$83.3 - 58.33 = 24.97$`. Wir haben aber auch echt wenige Beobachtungen. Auf der anderen Seite ist es dann für ein agrarwissenschaftliches Experiment gra nicht mal so wenig.
Und hier sehen wir dann auch gleich das Problem mit der Funktion `check_heteroscedasticity()`. Wegen der geringen Fallzahl sagt die Funktion, dass alles okay ist mit den Varianzen und wir keine Varianzheterogenität vorliegen haben.
```{r}
check_heteroscedasticity(toy_fit)
```
Wenn wir uns aber mal die @fig-toy-1 anschauen sehen wir, dass wir auf keinen Fall Varianzhomogenität vorliegen haben. Die geringe Fallzahl von zwölf Beobachtungen je Gruppe ist zu klein, damit die Funktion `check_heteroscedasticity()` eine signifikanten Abweichung finden kann. Deshalb schaue ich mir immer die Abbildungen an.
```{r}
#| echo: true
#| message: false
#| label: fig-toy-1
#| fig-align: center
#| fig-height: 5
#| fig-width: 10
#| fig-cap: "Überprüfung der Varianzhomogeniutät des Modells `toy_fit` mit der Funktion `check_model()`. Wir sehen hier eine klare Varianzheterogenität in dem Modell."
check_model(toy_fit, check = c("homogeneity", "normality"))
```
Wir können hier auch den `coeftest()` für Varianzheterogenität rechnen, aber wir sind ja hier an den Gruppenvergleichen interessiert, so dass wir dann gleich zu `emmeans` weitergehen.
```{r}
coeftest(toy_fit, vcov. = vcovHC, type = "HC1")
```
Wie schon oben gezeigt, können wir dann einfach `emmeans()` nutzen um die Gruppenvergleiche zu rechnen. Auch hier müssen wir einmal angeben, dass wir einen paarweisen Vergleich rechnen wollen. Wir wollen alle Gruppen miteinander vergleichen. Dann noch die Option `vcov. = sandwich::vcovHAC` gewählt um für heterogene Varianzen zu adjustieren.
```{r}
em_obj <- toy_fit |>
emmeans(~ trt, method = "pairwise", vcov. = sandwich::vcovHAC)
em_obj
```
Auch hier sehen wir die mittlere Wahrscheinlichkeit infiziert zu sein in der Spalte `emmean`. Die Standardfehler `SE` sind für jede Gruppe unterschiedlich, die Adjustierung für die Varianzheterogenität hat geklappt. Dann kannst du noch die paarweisen Gruppenvergleiche über einen Kontrasttest dir wiedergeben lassen.
```{r}
em_obj |>
contrast(method = "pairwise", adjust = "none")
```
Oder aber du nutzt das *compact letter display*.
```{r}
em_obj |>
cld(Letters = letters, adjust = "none")
```
Da die Fallzahl sehr gering ist, können wir am Ende nur zeigen, dass sich die Kontrolle von der Behandlung `killAll` unterscheidet. Hätten wir mehr Fallzahl, dann könnten wir sicherlich auch zeigen, dass der Unterschied zwischen der Kontrolle zu der Behandlung `roundUp` in eine signifikante Richtung geht. So klein ist der Unterschied zwischen Kontrolle und `roundUp` mit 41.7% ja nicht.
::: callout-tip
## Anwendungsbeispiel: Beschädigter Mais nach der Ernte
Im folgenden Beispiel schauen wir uns nochmal ein praktische Auswertung von einem agrarwissenschaftlichen Beispiel mit Mais an. Wir haben uns in diesem Experiment verschiedene Arten `trt` von Ernteverfahren von Mais angeschaut. Dann haben wir nach vier Zeitpunkten bestimmt, ob der Mais durch das Ernetverfahren nachträglich beschädigt war. Die Beschädigung selber wurde dann etwas komplizierter mit einem Labortest festgestellt, aber wir schauen uns nur die Ausprägung ja/nein also $1/0$ als Outcome an. Durch einen Fehler im Labor müssen wir eine Kombination für den letzten Tag und der dritten Behandlung entfernen.
```{r}
maize_tbl <- read_excel("data/maize_rate.xlsx") |>
mutate(damaged = ifelse(time == "5d" & trt == 3, NA, damaged),
trt = factor(trt, labels = c("wenig", "mittel", "viel")))
```
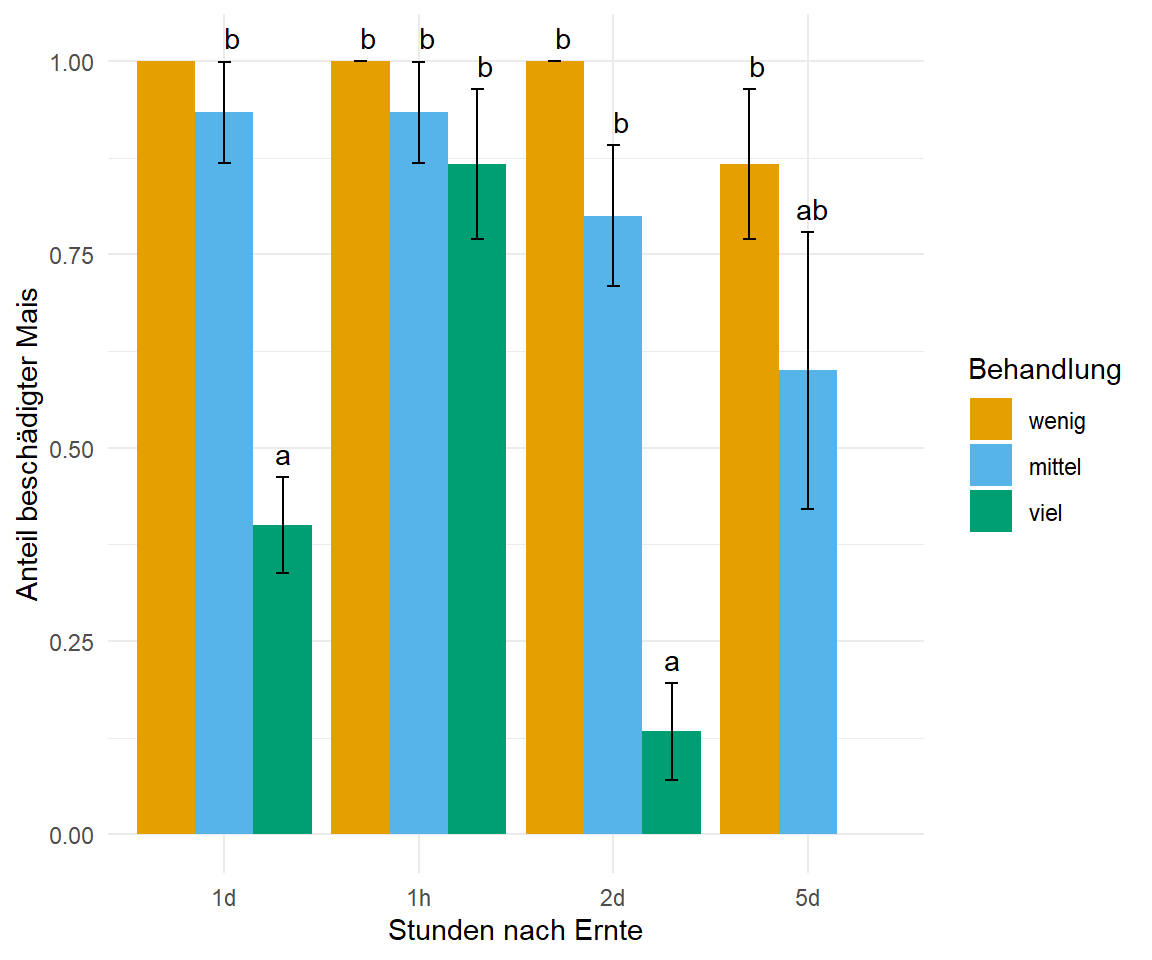
Dann rechnen wir auch schon das `lm()` Modell und nutzen `{emmeans}` für den Gruppenvergleich. Hier unbedingt `SE` als den Standardfehler für die Fehlerbalken nutzen, da wir sonst Fehlerbalken größer und kleiner als $0/1$ erhalten, wenn wir die Standardabweichung nutzen würden. Du solltest auch immer von Varianzheterogenität ausgehen, deshalb nutze ich hier auch die Option `vcov. = sandwich::vcovHAC` in `emmeans()`. In der @fig-lm-mod-prob-maize siehst du das Ergebnis der Auswertung in einem Säulendiagramm. Wir sehen einen klaren Effekt der Behandlung `viel`. Schade, dass wir dann nach 5 Tagen leider keine Auswertung für die dritte Behandlung vorliegen haben. Aber sowas passiert dann unter echten Bedingungen mal schnell.
```{r}
#| echo: true
#| warning: false
#| message: false
#| label: fig-lm-mod-prob-maize
#| fig-align: center
#| fig-height: 5
#| fig-width: 6
#| fig-cap: "Säulendigramm der Anteile des beschädigten Mais aus einem linearen Modell. Das `lm()`-Modell berechnet die Mittelwerte in jeder Faktorkombination, was dann die Anteile des beschädigten Mais entspricht. Das *compact letter display* wird dann in `{emmeans}` generiert."
lm(damaged ~ trt + time + trt:time, data = maize_tbl) |>
emmeans(~ trt * time, vcov. = sandwich::vcovHAC) |>
cld(Letters = letters, adjust = "bonferroni") |>
as_tibble() |>
ggplot(aes(x = time, y = emmean, fill = trt)) +
theme_minimal() +
labs(y = "Anteil beschädigter Mais", x = "Stunden nach Ernte",
fill = "Behandlung") +
geom_bar(stat = "identity",
position = position_dodge(width = 0.9, preserve = "single")) +
geom_text(aes(label = .group, y = emmean + SE + 0.01),
position = position_dodge(width = 0.9), vjust = -0.25) +
geom_errorbar(aes(ymin = emmean-SE, ymax = emmean+SE),
width = 0.2,
position = position_dodge(width = 0.9, preserve = "single")) +
scale_fill_okabeito()
```
:::